
В российской педиатрической практике диагноз цистиноза длительно оставался казуистическим, и пациенты, страдающие данным заболеванием, относились к группам тубулопатий неустановленного характера. Из-за незнания клиники не принимались во внимание глазные симптомы, эндокринные
нарушения и изменения со стороны центральной нервной системы (ЦНС), считавшиеся вторичными по отношению к почечной недостаточности. Прогрессирующее поражение почек заканчивалось терминальной почечной недостаточностью и в отсутствие возможности трансплантации – гибелью больных. Более того, часть больных погибали в раннем возрасте – задолго до наступления хронической почечной недостаточности (ХПН), вероятно вследствие дегидратации, тяжелых электролитных расстройств и ацидоза.
Опыт нашей работы с больными цистинозом непродолжителен, однако прогресс за последние пять лет очевиден и мы считаем возможным поделиться информацией о заболевании и текущем состоянии проблемы.
Классификация и эпидемиология
Цистиноз – редкое аутосомно-рецессивное заболевание, причиной которого является мутация в гене CTNS, кодирующем лизосомальный переносчик цистина. При этом заболевании в различных органах и тканях происходит накопление цистина в лизосомах и отложение его кристаллов. Выделено три клинические формы болезни, различающиеся по тяжести и времени дебюта. Наиболее тяжело протекает классическая инфантильная нефропатическая форма цистиноза (OMIM # 219800). Впервые она была описана в начале XX в. Aberhalden как заболевание, неизбежно приводящее к терминальной
уремии в детском возрасте [1, 2]. Позднее были опубликованы наблюдения и за более легкими случаями болезни. Ювенильная, или промежуточная, форма (OMIM #219900) также сопровождается поражением почек, но с манифестацией в подростковом возрасте. Взрослая, или “доброкачественная”,
форма цистиноза (OMIM # 219750) проявляется поражением глаз с развитием фотофобии без ризнаков вовлечения других органов. При этом варианте болезни кристаллы цистина откладываются только в роговице и костном мозге [3]. В США цистиноз встречается с частотой 1 : 200000; в Европе 1 : 179000; ювенильная и взрослая формы отмечаются менее чем в 5 % случаев.
Патофизиология и генетика
Клинические проявления при нефропатическом цистинозе также имеют определенный диапазон тяжести, но в среднем в отсутствие специфической терапии терминальная почечная недостаточность развивается к 10-летнему возрасту [4]. С развитием трансплантации почки продолжительность жизни
пациентов значительно увеличилась и в трансплантат болезнь не возвращается. Однако и после почечной трансплантации продолжается отложение кристаллов цистина в экстраренальных органах, включая мышцы, головной мозг, костный мозг, печень, селезенку, лимфатические узлы, роговицу, конъюнктиву, щитовидную, поджелудочную железы, яички и кишечник [1]. Наряду с прежней, в основном симптоматической, терапией было разработано специфическое лечение, направленное на
коррекцию основного патогенетического механизма. Было обнаружено, что длительная и упорная терапия цистеамином, донатором сульфгидрильных групп, вызывает деплецию цистина и значительно отдаляет почечную недостаточность, улучшает показатели роста ребенка и предотвращает серьезные поздние экстраренальные осложнения [7–16].
Ген цистинозина, CTNS, располагается на хромосоме 17p13 [17]. CTNS состоит из 12 экзонов и кодирует синтез цистинозина – интегрального белка лизосомальной мембраны, представленного 367 аминокислотами. Функцией цистинозина является перенос цистина из лизосомы в цитоплазму [18, 19].
В Северной Америке и Европе почти 75 % всех исследованных аллелей имеют делецию 57-kb, и это самая частая мутация CTNS [20]. Было описано около 60 других мутаций [21–24]. Пациенты с классическим инфантильным цистинозом имеют делеции или другие мутации, приводящие к полному отсутствию цистинозина, в то время как более легкие случаи (ювенильная и взрослая формы) обычно компаунд гетерозиготны и имеют наряду с тяжелой (например, нонсенс) более легкую мутацию (например, миссенс) [25]. У бессимптомных гетерозиготных носителей отмечается двукратное снижение активности цистинозина [26]. При этом уровень внутриклеточного цистина остается лишь слегка повышенным [27]. В целом, тяжесть клинических проявлений болезни, как правило, имеет пря-
мую корреляцию с концентрацией внутрилизосомального цистина и обратную корреляцию с резидуальной функцией цистинозина [28, 29]. Тем не менее нельзя исключать возможного модифицирующего влияния на фенотип других генов и экологических воздействий.
Самым ярким проявлением нарушения лизосомального транспорта цистина является образование кристаллов вследствие плохой растворимости цистина в воде. Кристаллы обычно имеют форму шестиугольника или прямоугольника, но могут быть игольчатыми и обладают свойством двойного преломления в поляризационном свете. Интересно, что тяжесть повреждения тканей не вполне коррелирует с распространенностью отложения кристаллов. Например, в печени и тонком кишечнике отмечается значительное интрацеллюлярное отложение кристаллов цистина, но это редко клинически проявляется. Этот факт, по всей вероятности, объясняется значительным функциональным резервом некоторых органов/более коротким клеточным циклом и быстрой сменой клеточной популяции. В некоторых клетках, например фибробластах, кристаллы никогда не образуются, несмотря на высокое содержание внутриклеточного цистина [30].
Клиническая картина
Симптомы цистиноза развиваются постепенно. Как правило, в первые 3–6 месяцев жизни клинические проявления болезни отсутствуют. К 6–12 месяцам появляются такие неспецифические симптомы, как анорексия, рвота, полиурия и запоры. Возможно развитие эпизодов лихорадки с признаками дегидратации. Существенно уменьшается прибавка в весе и росте, прогрессирующая в дальнейшем.
Поражение почек обычно манифестирует клиникой синдрома Фанкони (Де Тони–Дебре–Фанкони) к 1-му году жизни. У пациентов отмечается потеря жидкости и электролитов, аминоацидурия, низкомолекулярная протеинурия, нормогликемическая глюкозурия, фосфатурия, гиперкальциурия
и гиперхлоремический ацидоз. В дальнейшем отмечается прогрессирующая потеря гломерулярной функции и развитие хронической почечной недостаточности (ХПН) в 7–12 лет [30, 31]. В настоящее время цистиноз считается самой частой причиной врожденного синдрома Фанкони в детском
возрасте. Диурез может быть столь значительным, что у некоторых больных на начальном этапе предполагается наличие несахарного диабета [2]. У некоторых детей значительная экскреция кальция и фосфатов приводит к медуллярному нефрокальцинозу [32]. У многих, хотя и не у всех, пациентов развивается гипофосфатемический рахит с высокой экскрецией фосфатов, нормальным уровнем витамина D, повышением активности щелочной фосфатазы и такими клиническими
проявлениями, как остеомаляция, деформации костей и задержка формирования моторных навыков [9, 30]. При развитии гиперкальциурической гипокальциемии возможно возникновение тетании. Гипокалиемия (в ряде случаев ниже 2,0 mEq/l) может приводить к нарушениям сердечной проводимости. Карнитин также теряется при синдроме Фанкони [33], что может быть причиной мышечной слабости [34]. У взрослых больных наблюдается прогрессирующая потеря мышечной
массы, периферическая полинейропатия.
В Северной Америке и Европе цистиноз составляет приблизительно 5 % случаев ХПН у детей [31]. По данным The European Dialysis and Transplant Association Registry, средний возраст детей с цистинозом, нуждающихся в заместительной терапии, составляет 9,5 лет с колебаниями от 1 до 20 лет [35]. Гистопатологически при цистинозе развивается атрофия клеток проксимальных канальцев с их деформацией по типу лебединой шеи [36]. Весьма характерно повреждение подоцитов, приобретающих вид многоядерных гигантских клеток. В подоцитах и интерстициальных клетках отмечается отложение кристаллов цистина [35].
Характерно развитие полиэндокринопатии с нарушением функции щитовидной, поджелудочной, половых желез. Цистиноз поражает многие железы, в связи с чем у пациентов нарушаются потоотделение [5], продукция слезной жидкости, саливация, что делает их склонными к развитию гипертермии, сухости глаз, рвоте. У больных может отмечаться анемия, обусловленная прежде всего уменьшением ренальной продукции эритропоэтина при почечной недостаточности и в меньшей степени – отложением кристаллов цистина в костном мозге [37].
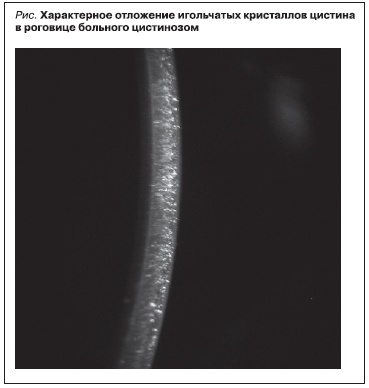
Ранними и патогномоничными проявлениями болезни являются отложения кристаллов в роговице и конъюнктиве [1]. Корнеальные кристаллы почти всегда имеются к возрасту 16 месяцев и выявляются при осмотре с помощью щелевой лампы в виде игольчатых, опалесцирующих помутнений (рис. 1) [4]. В первую очередь в этот процесс вовлекается эпителий роговицы. На второй декаде жизни развивается фотофобия и возможно возникновение тяжелой кератопатии с нарушением зрения. Иногда кристаллы откладываются и в передней камере, радужной оболочке, цилиарном теле, сосудистой оболочке, на
глазном дне и в зрительном нерве [16, 38]. В некоторых случаях развитие пигментной ретинопатии предшествует появлению корнеальных кристаллов [39].
Диагностика
Ранняя верификация диагноза и своевременное начало специфической терапии цистеамином имеют решающее значение для судьбы детей, страдающих цистинозом [10]. Диагноз цистиноза подтверждается определением высокого уровня цистина в гранулоцитах – отношение концентрации
цистина к концентрации белка более 0,5 нмоль/мг белка [2]. Для верификации цистиноза считается достаточным выявление кристаллов в роговице у пациента с синдромом Фанкони, т. к. подобные кристаллы не обнаруживаются при других причинах этого синдрома у детей, таких как тирозинемия, галактоземия, гепаторенальный гликогеноз, болезнь Дента, синдром Лове и т. д. С другой стороны, отсутствие кристаллической кератопатии не позволяет исключать цистиноз, особенно у младенцев. В прошлом для верификации диагноза применялась диагностическая биопсия различных органов, включая костный мозг, конъюнктиву и почки [2]. Однако в настоящее время общепризнанно, что для установления диагноза эти инвазивные процедуры не являются необходимыми. Ранний диагноз может быть установлен и с помощью молекулярно-генетического исследования. При наличии в семье хотя бы одного ребенка с цистинозом при каждой следующей беременности рекомендуется пренатальная диагностика болезни с помощью измерения уровня свободного цистина в культуре амниоцитов или
клетках ворсин хориона, или с помощью генетического исследования [2].
Терапия
Лечение цистиноза состоит из симптоматической и специфической терапии. Симптоматическая терапия направлена на коррекцию тубулярных потерь бикарбоната, воды, калия, натрия, фосфата под контролем их уровней в плазме [40]. Назначаются препараты витамина D для лечения рахита,
L-тироксин для коррекции гипотиреоза, L-карнитин до 50 мг/кг. При задержке роста – препараты рекомбинантного гормона роста.
Цистеамин (HS-CH2-NH2) – единственное вещество, доказавшее in vitro и in vivo свою эффективность в качестве супрессора отложения цистина внутри лизосом. Цистеамин проникает в лизосому, где расщепляет цистин на две молекулы цистеина и затем соединяется с одной из них с помощью дисульфидной связи [40, 41]. Образующиеся при этом цистеин-цистеаминовый комплекс и цистеин не нуждаются в цистинозине для выхода из лизосомы. Дисульфидное соединение, являющееся аналогом
лизина, переносится через лизиновый порт (рис. 1). Цистеамин производится в виде битартрата – коммерческое название препарата Цистагон (Cystagon®). Лечение должно начинаться сразу после подтверждения диагноза. Доза постепенно в течение 6–8 недель увеличивается от 0,2 до 1,3 грамма/м2 в день [2, 10]. Максимальная доза препарата составляет 1,95 грамма/м2 в день [9]. Цистеамин хорошо всасывается, его максимальный эффект развивается через 1–2 часа и длится не более 6 часов [5, 25]. Поэтому суточная доза должна назначаться в четыре приема. Поскольку ответ на терапию вариабельный, эффективность и достаточность дозы препарата контролируются определением концентрации цистина в гранулоцитах. Целевым значением через 6 часов после приема очередной дозы препарата является концентрация не выше 1 наномоль 1/2 цистина на миллиграмм белка [9]. Своевременная терапия цистеамином доказала свою эффективность в плане замедления снижения почечных функций и улучшения роста у детей с цистинозом в додиализную стадию [19]. Даже при позднем начале терапии возможно улучшение показателей клубочковой фильтрации, однако синдром Фанкони, как правило, не подвергается обратному развитию [28]. Оральная терапия цистеамином не приводит к растворению кристаллов в роговице. Этого эффекта можно достичь применением содержащих цистеамин глазных капель (0,5%-ный раствор – минимум 4 раза в день) [41].
Цистиноз в стадии терминальной уремии не представляет какой-либо специальной проблемы в плане выбора способа замещения функции почек. В этих обстоятельствах применяются и гемодиализ, и перитонеальный диализ, но методом выбора, как и при других вариантах конечной стадии болезни
почек в детском возрасте, является трансплантация почки [40]. В трансплантате никогда не рецидивирует синдром Фанкони, а выявление кристаллов цистина в почечной ткани объясняется их заносом лейкоцитами и макрофагами хозяина [1]. Родственники пациента, являющиеся гетерозиготными носителями цистиноза, могут быть донорами почки, т. к. это заболевание у них никогда не развивается.
Лечение цистеамином после трансплантации почки продолжается для воздействия на экстраренальные проявления цистиноза.
Собственные наблюдения
В настоящее время диагноз нефропатического цистиноза в России подтвержден у 8 пациентов в возрасте от 1,5 до 26 лет. Четырем из них диагноз был установлен в раннем детском возрасте на основании сочетания синдрома Фанкони и характерных данных офтальмологического исследования. У одного ребенка глазные изменения не выявлены и диагноз цистиноза подтвержден молекулярно-генетическим исследованием. В двух семьях имелись указания на гибель предыдущих детей в раннем возрасте при симптомах, сходных с имеющимися у детей, наблюдаемых нами.
У одного ребенка заболевание проявлялось протеинурией до 2 г в сутки. В возрасте 2 лет была выполнена нефробиопсия, показавшая фокально-сегментарный гломерулосклероз. Был назначен эналаприл, однако ребенок активно не наблюдался и попал в наше поле зрения лишь в возрасте 12 лет с картиной терминальной стадии ХПН. В то же время обнаружены характерные отложения игольчатых кристаллов в роговице, подтвердившие цистиноз. Была выполнена трансплантация
почки от живого родственного донора. На протяжении 3 лет функция трансплантата хорошая. У одной пациентки диагноз цистиноза был установлен лишь в возрасте 23 лет – через 7 лет после трансплантации почки, на основании специфического тяжелого кератоконъюнктивита и анамнестических указаний на “тубулопатию” как первичное заболевание почек.
У 7 детей диагноз был подтвержден молекулярно-генетическим исследованием, в большинстве случаев обнаружившим делеции в гене цистинозина, реже – миссенс и сплайсинг-мутации.
Лишь у троих детей, у которых диагноз цистиноза был установлен в возрасте до двух лет, одновременно было начато лечение Цистагоном®, на фоне которого уменьшилась полиурия.
Только у них на протяжении 2–3 лет к настоящему моменту остается сохранной скорость клубочковой фильтрации (СКФ). У оставшихся троих детей наблюдается прогрессирующее падение СКФ до 23–55 мл/мин/1,73 м2. Прогрессирование достигнуто только в уменьшении выраженности метаболических
изменений, связанных с синдромом Фанкони и рахита. Данный факт согласуется с общепринятой точкой зрения о том, что отсрочить развитие терминальной ХПН при цистинозе можно только ранним началом патогенетической терапии [41].
К сожалению, из-за отсутствия регистрации Цистагона® в Российской Федерации и нерегулярного снабжения препаратом эффективность лечения недостаточна. Недостаточной остается и настороженность врачей в отношении данного заболевания, что ведет к поздней диагностике или вовсе к отсутствию таковой. Решение данных проблем зависит от координированных действий медицинской общественности и коррекции законодательства в области орфанных заболеваний.


