
Введение
Исследования последних лет позволили установить связь между развитием тромбозов различной локализации и генетически обусловленной тромбофилией в отсутствие традиционных факторов риска. Вероятность формирования гиперкоагуляционного состояния возрастает с числом выявленных «прокоагулянтных» генотипов. При ГУС развитие микроциркуляторных тромбозов служит основным клиническим проявлением заболевания и поражение почек, как правило, сочетается с тромботической микроангиопатией сосудов головного мозга, легких, кишечника, печени, сердца, приводя к развитию полиорганной недостаточности с нарушением витальных функций. Роль тромбофилии в развитии патологии человека хорошо изучена в акушерской практике, неврологии, кардиологии, нефрологии [1–3]. Возможность поражения сосудов микроциркуляторного русла с развитием тромботической микроангиопатии была четко установлена при антифосфолипидном синдроме. Недавние исследования продемонстрировали развитие нефропатии у больных с «протромбогенными генотипами» генов MTHFR C677T, PAI-1 -675 4G/5G, FGB-455 G/A, ITGB3 L33P и доказали, что наличие мультигенной тромбофилии способствует более быстрому прогрессированию гломерулонефритов [1]. До настоящего времени роль наследственной тромбофилии в развитии и прогрессировании тромботической микроангиопатии у пациентов с ГУС, при котором почки являются главным органом-мишенью для тромбоза сосудов малого калибра, до сих пор не изучена. Комплексное изучение ассоциаций генетических маркеров тромбофилии, клинических особенностей ГУС может расширить представление о прогностических факторах, отдельных патогенетических механизмах, особенностях течения и подходах к лечению данного синдрома.
 Целью настоящего исследования стало изучение влияния генетически обусловленной тромбофилии на характер течения ГУС у детей.
Целью настоящего исследования стало изучение влияния генетически обусловленной тромбофилии на характер течения ГУС у детей.
Материал и методы
В исследование включены 47 пациентов с типичной формой ГУС в возрасте от 5 до 62 месяцев (19,3±2,1 месяца), из которых в возрасте от года до 3 лет были 59,6% (n=28); до года – 27,7% (n=13) и старше 3 лет – 12,8% (n=6) детей. Среди пациентов преобладали мальчики (66%, n=31).
Обследование включило стандартный набор клинико-лабораторных тестов, применяемых в отношении всех больных острой почечной недостаточностью. Всем больным исследовался аллельный полиморфизм шести различных генов системы свертывания крови: метилентетрагидрофолатредуктаза (MTHFR С677Т); ген протромбина (PTGG20210A); V фактора свертывания крови (FVLeidenG1691A); фибриногена (FGBG455A); тромбоцитарного рецептора фибриногена ITGB3 T176CL33P и ингибитор активатора плазминогена (PAI-1 4G/5G 675).
При анализе аллельных полиморфизмов генов выявлен нормальный генотип – «дикий», когда отсутствовали замены, гетерозиготный – при замене 1-го аллеля, либо гомозиготный – при замене 2 аллелей.
Методика исследования ДНК-полиморфизмов представлена межклинической лабораторией молекулярных методов диагностики Первого МГМУ им. И.М. Сеченова (зав. лаб. к.м.н. Е.М. Пальцева).
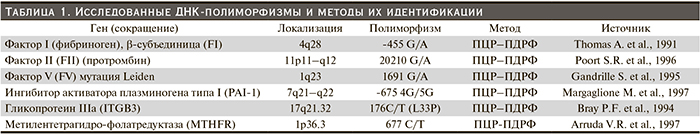
Для определения полиморфных аллелей генов MTHFR С677Т, PTGG20210A, FVLeidenG1691A, FGBG455A, ITGB3 T176CL33P использован метод полиморфизма длины рестриктивных фрагментов (ПЦР-ПДРФ); а гена PAI-1 4G/5G 675 – метод однонитевого конформационного полиморфизма ДНК (ПЦР- SSCP) (табл. 1).
Статистический анализ проведен на персональном компьютере с использованием статистического пакета SPSS-21.0. Посредством сравнения средних величин с использованием критерия Стьюдента проведена оценка параметрических данных. Правильность распределения событий в группах оценивалась с помощью сравнения с нулевой гипотезой. Во всех группах распределение было правильным и значимо не отличалось от нулевой гипотезы. При р<0,05 разница в группах считалась достоверной.
Результаты
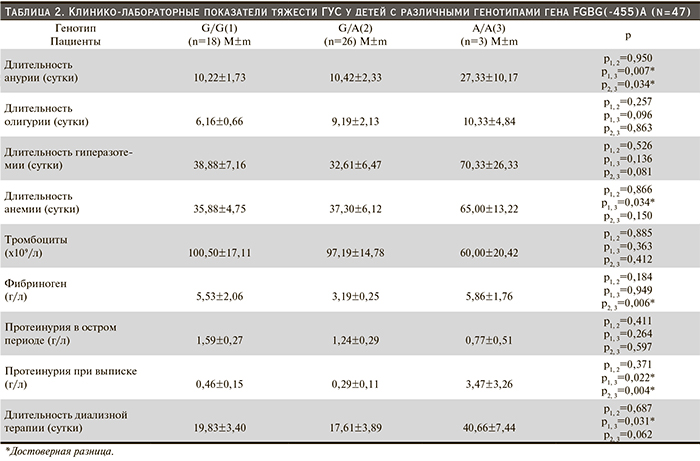
Среди 47 пациентов с ГУС 18 (38,3%) имели преобладающий в популяции генотип (G/G) («дикий») гена β-цепи фибриногена (FGBG(455)A), 26 (55,3%) пациентов обладали гетерозиготным генотипом (G/A) и лишь 3 (6,4%) детей оказались носителями гомозиготного (A/A-«мутантного») генотипа (рис. 1).
 Сравнение длительности анурии у больных с различными генотипами гена фибриногена показало, что у пациентов с носительством генотипа A/A значение этого показателя достоверно превосходило таковое у больных, имеющих «дикий» и гетерозиготный генотипы (р=0,007 и р=0,034). Длительность анемии у детей с генотипами G/G и G/A была соизмеримой и почти в 2 раза меньшей по сравнению с носителями генотипа А/А. Дети с мутантным генотипом А/А имели более выраженную тромбоцитопению по сравнению с носителями генотипов G/G и G/A (табл. 2).
Сравнение длительности анурии у больных с различными генотипами гена фибриногена показало, что у пациентов с носительством генотипа A/A значение этого показателя достоверно превосходило таковое у больных, имеющих «дикий» и гетерозиготный генотипы (р=0,007 и р=0,034). Длительность анемии у детей с генотипами G/G и G/A была соизмеримой и почти в 2 раза меньшей по сравнению с носителями генотипа А/А. Дети с мутантным генотипом А/А имели более выраженную тромбоцитопению по сравнению с носителями генотипов G/G и G/A (табл. 2).
У больных с генотипом А/А средняя продолжительность гиперазотемии почти вдвое превышала таковую при двух других генотипах, составив 70,3±26,3 суток. При генотипе А/А выявлена наибольшая длительность олигурии по сравнению с носителями «дикого» и гетерозиготного генотипов. Длительность диализной терапии пациентов с генотипом А/А вдвое превосходила таковую больных с генотипами G/G и G/A (р=0,03 и р=0,06).
В дебюте ГУС у детей с гетерозиготным генотипом гена FGBG(455) A G/A уровень фибриногена соответствовал норме, несмотря на более частое носительство аллеля А (доля аллеля – 0,28). Умеренная гиперфибриногенемия была характерной для пациентов с G/G- и А/А-генотипами.
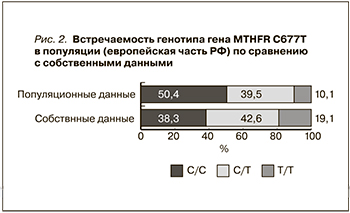
Суммарная частота встречаемости гетерозиготного (C/T) и мутантного (T/T) генотипов гена MTHFR составила 61,7%, что превышает данные в популяции, причем больше за счет Т/Т-генотипа (19,1 vs 10,1% соответственно) (рис. 2).
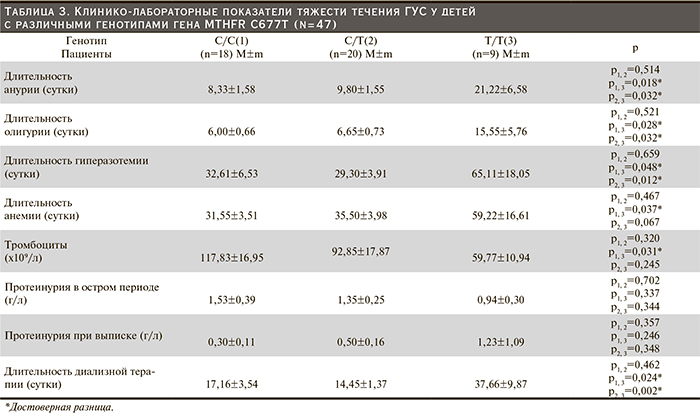
У пациентов с генотипом Т/Т длительность анурии более чем в 2 раза превышала таковую у больных с другими полиморфными вариантами (С/С и С/Т). Средняя длительность анемии у пациентов с этими генотипами была практически равной, тогда как при «мутантном» генотипе (Т/Т) продолжительность анемии оказалась почти вдвое большей, достоверно превосходя таковую у пациентов с генотипом С/С (р=0,037). При наличии последнего генотипа отмечена также минимальная выраженность тромбоцитопении по сравнению с генотипами С/Т и особенно Т/Т, носители которого имели выраженную тромбоцитопению. Отсутствие достоверности изученного показателя, по-видимому, можно объяснить недостаточной численностью больных. Пациенты с генотипом С/С и С/Т получали диализную терапию в течение значительно более короткого срока, чем больные с носительством генотипа Т/Т.
У детей с генотипом С/С и С/Т длительность олигурии была сопоставимой и более чем в 2 раза меньшей, чем при генотипе Т/Т. Аналогичная закономерность отмечена также в отношении продолжительности гиперазотемии.
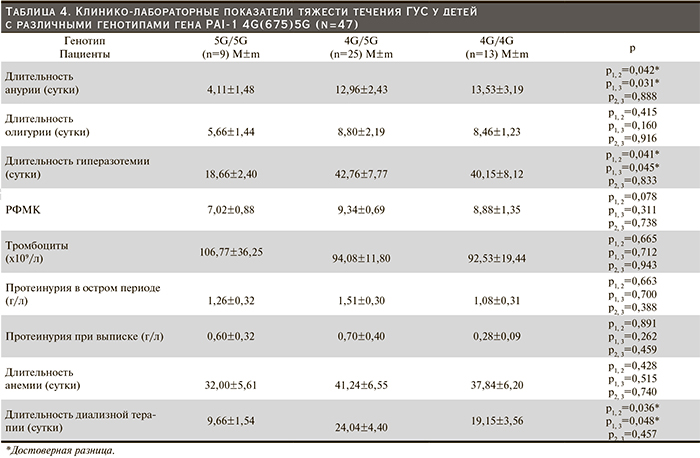
Среди 47 наблюдаемых 9 (19,2%) пациентов имели нормальный генотип 5G/5G гена PAI-1 4G(675)5G, 25 (53,2%) являлись носителями преобладающего в популяции гетерозиготного генотипа 4G/5G и 13 (27,6%) – носителями гомозиготного генотипа 4G/4G (рис. 3). Суммарная частота гетерозиготных и «мутантных» генотипов гена PAI-1 была выше, чем в популяции (80,8% vs 63,2% соответственно), причем за счет большего преобладания пациентов с 4G/4G-генотипом.
Число тромбоцитов у детей с различными генотипами гена PAI-1 практически не различалось. Длительность анемии у пациентов с «протромбогенными» генотипами недостоверно превосходила таковую у пациентов с «диким» полиморфным вариантом.
Значимая разница длительности нарушений азотовыделительной функции почек выявлена у пациентов с генотипом 4G/5G и 4G/4G в отличие от детей, имеющих нормальный генотип, что, по-видимому, объясняется тяжестью почечного повреждения у носителей аллеля 4G.
У детей с генотипами 4G/5G и 4G/4G длительность диализной терапии достоверно превосходила таковую у пациентов, обладающих нормальным генотипом, что полностью соответствовало длительности у них периода гиперазотемии. Продолжительность олигурии у детей с гетерозиготным и гомозиготным генотипами была недостоверно выше по сравнению с носительством «дикого» генотипа.

Анализ средних показателей РФМК в зависимости от генотипа гена PAI-1 не выявил достоверных различий в группах обследуемых и во всех случаях превышал референсные значения.
Средний уровень протеинурии в острую стадию болезни мало отличался у детей с разными генотипами. При выписке у детей с генотипом 4G/4G протеинурия была наименьшей, хотя достоверно не отличалась от таковой у детей с генотипом 5G/5G и 4G/5G.
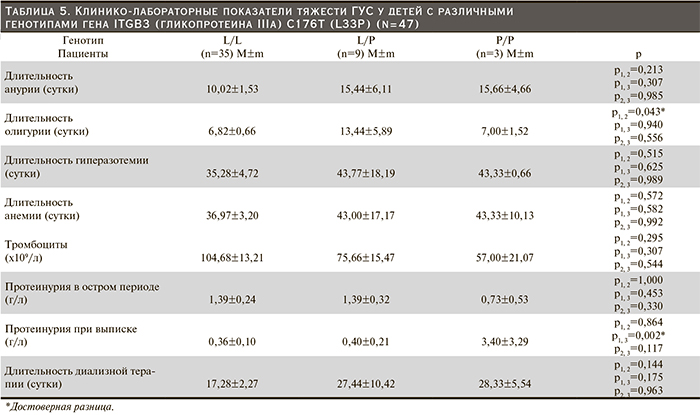
При анализе полиморфных вариантов гена тромбоцитарного рецептора фибриногена (ITGB3 [гликопротеина IIIa] С176Т [L33P]) обнаружено, что у 74,5% (n=35) пациентов имел место «дикий» генотип L/L гена, 19,1% (n=9) пациентов обладали гетерозиготным (L/P) и 6,4% (n=3) являлись носителями гомозиготного генотипа (P/P) (рис. 4).
Преимущественное большинство пациентов в нашем исследовании имели «дикий» генотип, что совпадает с популяционными данными. Однако гетерозиготные генотипы встречались в 1,7 раза реже, а «мутантные» (гомозиготные) более чем в 7 раз чаще, чем в популяции.
У пациентов с гомозиготой по аллелю L длительность анурии была недостоверно меньше, чем у лиц с гетерозиготным и гомозиготным генотипами (табл. 5).
При гетерозиготном и гомозиготном генотипах продолжительность анемии была практически одинаковой, недостоверно превосходя таковую у пациентов с «диким» генотипом. Среднее число тромбоцитов было наименьшим у детей с Р/Р-генотипом. При генотипе L/L уровень тромбоцитов был недостоверно выше, чем у детей с генотипом L/P.
Длительность олигурии была сравнимой у детей с генотипами L/L и Р/Р и, напротив, у детей с гетерозиготным генотипом L/P оказалась почти вдвое больше по сравнению с носителями упомянутых полиморфных форм гена ITGB3. Пациенты с генотипом L/P и Р/Р имели одинаковую длительность гиперазотемии, оказавшейся несколько выше, чем у носителей «дикого» генотипа.
У пациентов с «диким» генотипом полиморфизма (L/L) уровень протеинурии в остром периоде практически совпадал с величиной протеинурии при гетерозиготном генотипе. Среднее значение протеинурии у детей с генотипом P/P было недостоверно меньше. При разрешении острой почечной недостаточности у детей с генотипом L/L и L/P уровень протеинурии был соизмеримым и почти в 4 раза ниже по сравнению с острым периодом. У детей с «мутантным» генотипом средняя величина протеинурии были на порядок выше.
Длительность диализной терапии пациентов с «протромбогенными» генотипами (L/P и P/P) оказалась в 1,5 раза выше, чем у детей, имевших «дикий» генотип L/L. Полученные данные подкрепляются данными о продолжительности гиперазотемии, которая также была меньше у детей с генотипом L/L.
Среди 47 пациентов с ГУС в 93,6% (n=44) случаев преобладающим оказался «дикий» генотип (G/G) гена протромбина (PTG G20210 A) (рис. 5). Лишь 6,4% (n=3) пациентов имели гетерозиготный генотип (G/A) этого гена, что в 3 раза превышало популяционные данные. Носителей гомозиготного генотипа (A/A) среди пациентов с ГУС выявлено на было, как и среди здоровых лиц.
У пациентов с носительством нормального генотипа (G/G) длительность анурии оказалась более высокой, чем у пациентов с гетерозиготным генотипом, однако эти данные недостоверны в связи с малым числом пациентов с генотипом G/A (табл. 6).
Средняя длительность олигурии, гиперазотемии у детей с выявленными генотипами практически не различалась. У пациентов с генотипом G/A длительность анемии, выраженность тромбоцитопении были недостоверно выше по сравнению с обладателями генотипа G/G. Достоверно выше у носителей гетерозиготного генотипа оказалась выраженность протеинурии по сравнению с обладателями гомозиготы по аллелю G. В то же время длительность диализной поддержки у пациентов с генотипом G/G была недостоверно больше, чем у детей с генотипом G/A, что, по-видимому, объясняется малой выборкой пациентов с носительством G/A.
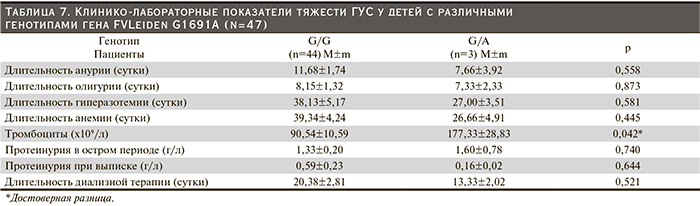
Среди 47 пациентов с ГУС 44 (93,6%) имели также преобладающий в популяции «дикий» генотип G/G гена FV Leiden G1691A, а 3 (6,4%) – гетерозиготный, что в 1,5 раза превышало данную частоту среди здоровых лиц. Как в популяции, так и в изучаемой группе ни в одном случае не было выявлено носителей гомозиготного генотипа (рис 6).
У пациентов с генотипом G/G продолжительность анурии была недостоверно выше по сравнению с больными – носителями генотипа G/A, что объясняется малой выборкой и большим разбросом данных (табл. 7). У детей с генотипами G/G и G/A средняя длительность олигурии была близкой по значению. Средняя продолжительность гиперазотемии, анемии, диализной поддержки у пациентов с генотипом G/G была больше, чем у детей с гетерозиготным генотипом. Напротив, число тромбоцитов при носительстве «дикого» генотипа G/G оказалось достоверно наименьшим. Выраженность протеинурии в остром периоде была практически одинаковой при обоих изученных генотипах, а при восстановлении диуреза оказалась большей у детей с генотипом G/G.
Обсуждение
У детей с ГУС преобладают «протромбогенные» генотипы гена FGB G(455)A, а наличие «мутантного» генотипа (А/А) данного гена значительно утяжеляет течение заболевания, проявляющееся удлинением периода острой почечной недостаточности и замедлением восстановления функции почек. По-видимому, это обусловлено избыточным (по сравнению с пациентами – носителями «дикого» или гетерозиготного генотипа) тромбообразованием в микроциркуляторном русле почек, приводящим к более выраженной ишемии органа. Похожие результаты, но касающиеся сосудов головного мозга были получены недавно M. Martiskainen et al., установившими, что наличие аллеля А служит фактором риска ишемического инсульта [4].

Согласно полученным данным, тяжесть клинических проявлений ГУС не различалась среди пациентов с «диким» и гетерозиготным генотипами гена MTHFR С677Т. Наличие мутантного полиморфизма гена MTHFR С677Т оказалось фактором риска развития тяжелой формы ГУС и, по-видимому, может рассматриваться как маркер неблагоприятного прогноза, что, скорее всего, связано с повреждающим действием гипергомоцистеинемии на эндотелий сосудов микроциркуляторного русла почек [5–6]. Наши данные сопоставимы с результатами исследования других видов почечной патологии в зависимости от генотипа гена MTHFR. Так, C. Zou et al. [7] доказали, что раннее развитие и прогрессирование ФСГС у детей может быть ассоциировано с Т/Т-генотипом, а O. Liangos et al. [8] обнаружили, что носительство данного полиморфного варианта может быть ассоциировано с ранним отторжением трансплантированной почки. Более того, C. Sucker et al. [9] рассматривают генотип Т/Т гена MTHFR как фактор риска развития ТМА (ГУС/ТТП), в первую очередь ОПН как наиболее яркого ее проявления. Возможной причиной служит гипергомоцистеинемия, развивающаяся у больных с мутантным генотипом гена MTHFR, что индуцирует развитие эндотелиальной дисфункции, и без того имеющей место у больных с тромботической микроангиопатией независимо от ее генеза.
Носительство гетеро- и гомозиготных генотипов гена PAI-1 4G(675)5G влияет на тяжесть почечного повреждения (длительность анурии, гиперазотемии, диализной поддержки, восстановления почечных функций). У детей с ГУС, по-видимому, имеет место неадекватный фибринолиз, обусловленный недостаточным образованием активного плазмина из-за преобладания «тромбофилических» генотипов (4G/5G и 4G/4G) гена PAI-1, связанных с повышенной выработкой ингибитора активатора плазминогена I типа [10–13].

Генетически детерминированный избыток PAI-1, по современным представлениям, не только ассоциирован с высоким риском сердечно-сосудистых катастроф (острый инфаркт миокарда, острое нарушение мозгового кровообращения), но и, по-видимому, играет важную роль в прогрессировании нефропатий, поскольку, как было установлено G. Grandaliano et al. [14], концентрация PAI-1 в ткани почек прямо коррелирует с процессами фиброза. Недавними исследованиями A.Y. Wang et al. [15] было показано, что 4G/4G генотип PAI-1 ассоциирован с активностью волчаночного нефрита и, как установили H. Suzuki et al., с прогрессированием IgA-нефропатии [16]. Разными исследователями изучалось не только влияние моногенного полиморфизма гена PAI-1 на почечное повреждение, но и мультигенного полиморфизма на макроорганизм. Так, С.И. Капустин [17] и S.J. Sheereen et al. [18] продемонстрировали, что сочетание данного полиморфизма гена PAI-1 с мутацией фактора FV Leiden, протромбина G20210A, MTHFR C677T повышает риск развития венозных тромбозов, что может иметь значение для развития тромбоза микроциркуляторного русла в период острых проявлений ГУС.
«Протромбогенное» носительство гена ITGB3 ассоциировано с тяжестью гематологических проявлений тромботической микроангиопатии при ГУС. Носительство гомозиготной мутации данного гена определяет большую выраженность тромбоцитопении, что может быть обусловлено более активным потреблением тромбоцитов в процессах микроциркуляторного тромбообразования вследствие повышенной склонности их к агрегации носителей аллеля Р. Это предположение косвенно подтверждают данные исследования, установившего связь полиморфизма ITGB3 с синдромами иммунных разрушений тромбоцитов, особенно с неонатальной тромбоцитопенией и аллоиммунной посттрансфузионной пурпурой [19].
Наличие гетерозиготного генотипа генов PTGG 20210 A и FVLeiden G1691A у пациентов с ГУС, вероятно, не влияет на тяжесть клинических проявлений заболевания, что может объясняться малой выборкой пациентов с данным генотипом.
Таким образом, в результате исследования заметно отчетливое преобладание «протромбогенных» генотипов всех исследуемых генов в группе пациентов с ГУС по сравнению со здоровой российской популяцией. Так, в 2 раза чаще встречался «протромбогенный» гомозиготный генотип генов MTHFR и ITGB; существенно выше – гетерозиготный генотип генов FGB и PAI-1. У носителей гетеро- и особенно гомозиготных генотипов генов MTHFR, ITGB, FGB и PAI-1 отмечена большая тяжесть клинических проявлений ГУС (длительность анемии, анурии, гиперазотемии, восстановления почечных функций, выраженность тромбоцитопении, продолжительность диализной терапии), что может определять отдаленный прогноз пациентов.
Полученные результаты свидетельствуют о том, что наследственная тромбофилия у детей с ГУС не служит фактором, обеспечивающим индукцию и генерализацию микроциркуляторного тромбообразования. Однако ассоциации генетических маркеров тромбофилии могут определять предрасположенность к развитию гиперкоагуляционного состояния, осложняющего течение болезни и, следовательно, усугубляющего его тяжесть, что, по-видимому, следует рассматривать как неблагоприятный прогностический фактор. Однако это нуждается в дальнейшем изучении.
Проведенный анализ «протромбогенных» генотипов дает основание предполагать, что микроциркуляторные тромбозы при ГУС могут развиваться при сочетании различных механизмов, связанных с генетически обусловленными дефектами в плазменном звене коагуляции (β-цепь фибриногена), системе фибринолиза (PAI-1), а также эндотелиальной дисфункцией (MTHFR).
Специфичность влияния «протромботических» генотипов на функциональную активность системы гемостаза, по-видимому, способна определять локализацию тромботического процесса, клиническую гетерогенность, а также прогноз тромботических осложнений. Дети с ГУС и выявленной наследственной тромбофилией нуждаются в тщательном наблюдении из-за возможности развития рецидивирующих тромбозов в микроциркуляторном русле почек под воздействием дополнительных факторов риска тромбообразования [20–23].


