
Введение
Мембранопролиферативный гломерулонефрит (МПГН) до недавнего времени рассматривался как вариант иммунокомплексного нефрита, клинически проявляющегося гематурией, протеинурией различной степени выраженности, нефротическим синдромом, нередко в сочетании с элементами остронефритического синдрома, в т.ч. (в ряде случаев) нарушением функции почек [1].
По происхождению выделяли первичный, или идиопатический, МПГН, этиология которого оставалась неясной, и вторичный МПГН с установленной этиологией [2]. Идиопатический МПГН, как правило, дебютирует в детстве мочевым синдромом (порой слабовыраженным), который может длительно персистировать, однако у части больных изменения постепенно нарастают, приводя к формированию нефротического и/или остронефритического синдромов. Причины вторичного МПГН многообразны и включают системную патологию (СКВ, синдром Шегрена, ревматоидный артрит), инфекции (в первую очередь гепатиты С и В, а также др. вирусные, бактериальные, грибковые и паразитарные заболевания [1]), моноклональную гаммапатию [1]. Поскольку причину развития МПГН удавалось установить далеко не всегда, в недавнем прошлом идиопатическая форма болезни была доминирующей. Диагноз устанавливался при светооптической микроскопии на основании выраженной мезангиальной пролиферации и утолщения гломерулярной базальной мембраны (ГБМ), нередко с двойным контуром. Эти изменения определили название данной формы гломерулонефрита (ГН) – мембранопролиферативный, или мезангиокапиллярный [1, 3]. Исторически сложилось, что в Европе и в нашей стране чаще использовался термин «мезангиокапиллярный» ГН, а в Северной Америке – «мембранопролиферативный» [3]. Однако сегодня практически повсеместно принято использовать последний термин [4]. Наличие иммунных депозитов, выявляемых при электронной микроскопии, считается характерной чертой МПГН. По локализации депозитов выделяли три типа болезни. I тип характеризовался наличием депозитов в мезангиальной и субэндотелиальной областях (т.н. классический МПГН), II тип – электронно-плотными депозитами, располагавшимися преимущественно в интрамембранозной, а также мезангиальной областях [2, 5]. В связи с характерным видом этих очень плотных интрамембранозных электронных депозитов данный тип ГН с 1975 г. по предложению R. Habib стали называть болезнью плотных депозитов (dense deposit disease, DDD) [6]. III тип МПГН характеризовался комбинацией признаков первых двух типов: депозиты в мезангиальной, субэндотелиальной областях в сочетании с субэпителиальными и/или интрамембранозными депозитами [2]. Ряд авторов подразделяли III тип на два подтипа (с субэпителиальными депозитами – Burkholder субтип [7] и интрамембранозными – Strife and Anders субтип [8, 9]. Однако не всегда выявленные изменения укладывались в рамки какого-то одного типа и в одном биопсийном препарате, а порой даже в пределах одного клубочка, патолог мог видеть признаки сразу нескольких типов МПГН [5].
Таким образом, прежняя классификация МПГН основывалась на результатах световой (СМ) и электронной микроскопии (ЭМ) и давала представление о характере повреждения клубочков и локализации иммунных депозитов, но не о механизме развития данного повреждения. Это привело к трудностям с определением подходов к лечению: до настоящего времени эффективность терапии МПГН по сравнению с другими морфологическими вариантами относительно низка.
До недавнего времени патогенез всех типов МПГН представлялся иммунокомплексным с обязательной последующей активацией комплемента по классическому пути (чем объяснялось свечение иммуноглобулинов и компонентов комплемента в депозитах и снижение в крови общей гемолитической активности комплемента СН50 у некоторых пациентов), однако в ряде случаев патологи отмечали свечение только С3-компонента комплемента в отсутствие иммуноглобулинов (ИГ), что не укладывалось в стандартное представление об МПГН. Активное изучение роли альтернативного пути активации комплемента в патогенезе заболевания в последние годы привело к появлению нового представления о природе и патогенезе МПГН и обновлению классификации. В 2011 г. на основе данных иммунофлюоресцентной микроскопии (ИФ) (свечение ИГ и/или комплемента) S. Sethi and F. Fervenza предложили новую систему классификации, согласно которой МПГН может быть подразделен на 2 вида: опосредованный иммунными комплексами (ИК-опосредованный) и комплемент-опосредованный МПГН [10] (рис. 1). ИК-опосредованный МПГН развивается при высоком уровне циркулирующих иммунных комплексов (ИК), комплемент-опосредованный МПГН – вследствие нарушения регуляции альтернативного пути комплемента [1].
ИК-опосредованный МПГН
Развитие ИК-опосредованного нефрита обусловлено постоянным присутствием в крови антигена, что приводит к образованию и отложению в клубочках комплексов антиген–антитело при хронических инфекциях, аутоиммунных болезнях [1, 11]. ИК вызывают активацию классического пути комплемента (КПК) и отложение его компонентов в мезангии и вдоль капиллярных стенок (субэндотелиально), что находит отражение в свечении ИГ и компонентов комплемента, выявляемом при ИФ [1]. Таким образом, очевидно, что иммунокомплексный МПГН всегда вторичен по отношению к основному заболеванию, приведшему к развитию нефрита. Это могут быть аутоиммунные заболевания (СКВ, синдром Шегрена, ревматоидный артрит [1, 12]), инфекции (HCV-инфекция, нередко с криоглобулинемией [1, 12–15], HBV-инфекция [1, 12], бактериальный эндокардит [1, 12, 16], шунт-нефрит, грибковые и паразитарные инфекции [1, 12]), моноклональные гаммапатии (лимфома, множественная миелома, а также MGUS – моноклональная гаммапатия неопределенного значения [1, 17]) (рис. 1).
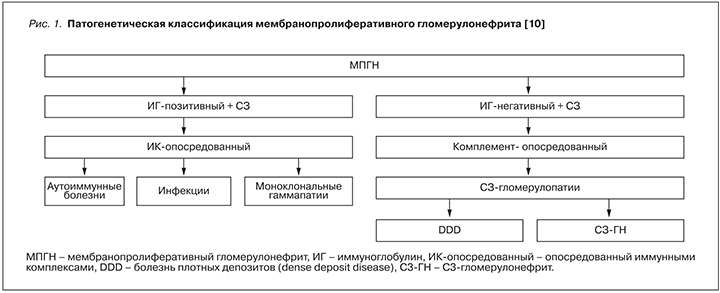
Некоторые авторы предлагают выделить отдельную группу МПГН, ассоциированного с моноклональной гаммапатией, вместо MGUS [17].
По локализации иммунных депозитов и их составу ИК-опосредованный гломерулонефрит может представлять собой МПГН I или III типов [11]. К особой группе МПГН вторичной этиологии можно отнести также тромботическую микроангиопатию (ТМА), имеющую при хроническом течении гистологические признаки МПГН на светооптическом уровне, но отличающуюся отсутствием свечения ИГ и компонентов комплемента при ИФ [18].
Комплемент-опосредованный МПГН
Комплемент-опосредованный МПГН связан с активацией альтернативного пути комплемента (АПК), приводящей к отложению его компонентов в мезангии и вдоль стенок капилляров без депозиции ИК [19]. Это доказано серией исследований почечной ткани пациентов методом лазерной микродиссекции с масс-спектрометрией, продемонстрировавших в большинстве случаев отложение в клубочках С3, С5 и С5b-9 без отложений ИГ у больных комплемент-опосредованным МПГН в отсутствие таковых у больных другими формами ГН и здоровых людей [18–21]. Аналогичные исследования были проведены в эксперименте [22, 23].
Система комплемента
Система комплемента является важнейшей частью врожденного неспецифического иммунитета, обеспечивающей первичную защиту организма от инфекционных агентов. Это система циркулирующих в кровеносном русле белков-энзимов, последовательно вступающих в реакции и активирующих друг друга (каскад ферментативных реакций расщепления). Активация комплемента вызывает мощный воспалительный ответ, реализующийся хемотаксисом (с участием анафилотоксинов С3а и С5а), опсонизацией (с участием опсонина C3b) и лизисом чужеродных клеток [1]. Основные этапы активации комплемента включают: 1) формирование С3-конвертазы – ферментативного комплекса, расщепляющего присутствующий в сыворотке С3; 2) формирование С5-конвертазы – ферментативного комплекса, расщепляющего С5-компонент и 3) сборку мембранатакующего комплекса (МАК) или терминального комплекса комплемента (ТКК), С5b-9, лизирующего чужеродную клетку. Комплемент активируется тремя путями: классическим, лектиновым и альтернативным. Различие состоит в начальном этапе процесса активации комплемента, но в дальнейшем все пути сходятся в центральном его звене – расщеплении С3-компонента, запускающем цепь реакций, приводящих в конечном счете к образованию МАК (рис. 2).

Классический путь инициируется связыванием С1q-компонента комплемента с комплексами антиген–антитело, лектиновый – маннозосвязывающего лектина (MBL), имеющего схожую структуру с С1q, с маннозными или глюкозаминовыми группами бактериальных полисахаридов. В дальнейшем это приводит к образованию С2а- и С4b-фрагментов, которые формируют С3-конвертазу классического пути (КП), С4bС2а, направленную на дальнейшее расщепление С3, центрального компонента всей системы комплемента. Образовавшаяся С3-конвертаза расщепляет С3 с образованием активированного С3b-фрагмента, присоединение которого к С3-конвертазе формирует С5-конвертазу, в свою очередь расщепляющую С5-компонент на анафилотоксин С5а- и С5b-фрагмент. Последний запускает сборку МАК (рис. 2).
Альтернативный путь активации комплемента является эволюционно более древним и инициируется непосредственно микроорганизмами (поверхностью клетки с полисахаридной структурой, например бактериальным липополисахаридом) без участия ИК. С1-, С2- и С4-компоненты не участвуют в активации АПК, и каскад реакций начинается с С3-компонента, который постоянно циркулирует в крови. АПК находится в состоянии постоянной активации, исходный уровень которой низок (т.н. механизм холостого хода), что обеспечивается спонтанным гидролизом свободно циркулирующего С3-компонента. Взаимодействуя с С3, вода вызывает медленный гидролиз тиоэфирной связи, повышая сродство С3 к присутствующему в сыворотке фактору В. Данная реакция приводит к образованию нестабильной сывороточной С3-конвертазы СЗbB. От нее под действием фактора D отщепляется фрагмент Ва, и образовавшаяся активная протеаза Bb формирует с СЗb стабильный ферментативный комплекс – С3-конвертазу альтернативного пути (АП), СЗbBb, функционально гомологичную С3-конвертазе КП. Расщепление С3 под действием сывороточной С3-конвертазы приводит к образованию фрагментов СЗb и С3а. При контакте с клеточной мембраной патогена или собственной клетки СЗb прикрепляется к ее поверхности и запускает дальнейшую сборку МАК. С3-конвертаза АП многократно усиливает расщепление С3 с образованием СЗb и формированием новых комплексов мембраносвязанной С3-конвертазы. Этот процесс получил название «петля амплификации» (усиления). При дальнейшем присоединении к С3-конвертазе дополнительного фрагмента СЗb образуется С5-конвертаза (СЗbBbСЗb), ферментный комплекс, расщепляющий С5-компонент с образованием мощного анафилотоксина С5а- и С5b-фрагмента, запускающего сборку МАК, С5b-9, который вызывает лизис бактериальных клеток [2, 24, 25] (рис. 2).
Активация и регуляция системы комплемента
Поверхность клеток хозяина в норме защищена от локальной амплификации и депозиции СЗb рядом регуляторных факторов комплемента, представленных как плазменными, так и мембраносвязанными белками, фиксированными на поверхности эндотелиальных клеток. Основными плазменными белками, регулирующими АПК, являются факторы Н (CFH) и I (CFI), мембраносвязанным – мембранный кофакторный протеин (MCP). Фактор H – основной регуляторный фактор АПК – блокирует образование С3-конвертазы и ускоряет ее распад, препятствуя процессу амплификации. Кроме того, фактор Н является кофактором CFI в инактивации С3b с образованием неактивного iС3b-фрагмента, не способного связаться с фактором В и образовывать С3-конвертазу. MCP связывает С3b и является дополнительным кофактором CFI. Таким образом, взаимодействие регуляторных факторов приводит к инактивации С3b, тем самым блокируя ключевой механизм активации – образование массы С3-конвертазы с последующей безудержной продукцией МАК (рис. 2).
Дисфункция АПК в жидкой фазе (циркуляции) приводит к отложению факторов комплемента в почечных клубочках с последующим повреждением и пролиферативным ответом, индуцируя развитие патологии, получившей название «С3-гломерулопатии». Напротив, бесконтрольная активация АПК на поверхности эндотелиальных клеток приводит к эндотелиальному повреждению с последующим развитием т.н. комплемент-опосредованной ТМА «атипичный гемолитико-уремический синдром» (аГУС) [26].
Дисрегуляция АПК может быть обусловлена врожденными (мутации в генах, кодирующих регуляторные белки) или приобретенными (образование аутоантител к этим белкам) дефектами [23, 27]. Известны мутации белков, регулирующих сборку и активность С3-конвертазы и инактивацию С3b. Основная часть мутаций регуляторных белков (CFH, CFI) приводит к их количественному или чаще функциональному дефициту, что нарушает нормальную регуляцию АПК, вызывая персистирующую неконтролируемую его активацию. Однако мутации генов С3-компонента и фактора В, напротив, усиливают функциональную активность этих белков, также способствуя избыточной активации АПК. Так, мутация гена, кодирующего С3-компонент, приводит к формированию мутантной С3-конвертазы, устойчивой к расщеплению фактором Н [27, 29]. Эффект, аналогичный последствиям этой мутации, имеют некоторые аутоантитела. Наиболее известным из них является С3-нефритический фактор (С3-Неф), представляющий собой аутоантитело к С3-конвертазе АП, которое стабилизирует этот комплекс, продлевая время его полужизни с нескольких секунд до 60 минут [30, 31], и препятствующее его инактивации фактором Н [24, 32]. Описаны также аутоантитела к факторам Н, I, антитела к компонентам С3-конвертазы – C3b и фактору В, приводящие к дисрегуляции АПК [19, 21, 23, 30, 32–35] (рис. 2).
Таким образом, комплемент активируется по альтернативному пути в результате спонтанного гидролиза С3-компонента и выходит из-под контроля вследствие дефектов в системе регуляции комплемента. Результатом неконтролируемой активации комплемента в циркуляции является последовательная активация его компонентов с их отложением в мезангиальной и субэндотелиальной областях, а также в капиллярной стенке, вызывающим локальное воспаление, пролиферацию с интерпозицией мезангиальных клеток в толщу ГБМ. Это приводит к ее расщеплению, и ГБМ приобретает характерный удвоенный вид («трамвайные пути»). ИГ не участвуют в данном процессе, поэтому комплемент-опосредованный МПГН, как правило, является ИГ-негативным и комплемент-положительным при ИФ. Расшифровка деталей патогенеза позволила выделить данный вид патологии в отдельную группу – С3-гломерулопатии.
С3-гломерулопатии
С3-гломерулопатии представляют собой группу редких заболеваний, связанных с дисрегуляцией АПК, включают главным образом болезнь плотных депозитов (DDD) и С3-гломерулонефрит (С3-ГН), а также CFHR5-нефропатию [22, 36]. Выделенные в 2011 г. в особую группу болезней в структуре МПГН С3-гломерулопатии (С3ГП) представляют собой заболевания, имеющие мембранопролиферативный вариант, «паттерн» повреждения, но иной (не иммунокомплексный) механизм развития, связанный с активацией АПК (комплемент-опосредованный МПГН) [37]. Соответственно, при ИФ микроскопии обнаруживается свечение только С3-компонента комплемента (допускается следовое содержание IgM/IgG), а при ЭМ – отложение его в мезангии и вдоль ГБМ субэндотелиально или интрамембранозно [18]. По составу и локализации отложений С3ГП разделяют на DDD, характеризующуюся электронно-плотными интрамембранозными депозитами, и С3-ГН с отложением С3 в мезангиальной, субэндотелиальной и/или субэпителиальной областях (но не интрамембранозно).
DDD чаще дебютирует в детском возрасте, С3-ГН чаще встречается у взрослых. Клиническая картина примерно одинакова и складывается из умеренного мочевого синдрома, включающего протеинурию субнефротического уровня, гематурию и в ряде случаев нарушения функции почек. DDD, как правило, носит спорадический характер, при С3-ГН возможны семейные случаи. Наиболее частой причиной DDD является наличие антител к С3-конвертазе (С3-Неф), а также мутации гена фактора H и антитела к нему, тогда как развитие С3-ГН, как правило, связано с наличием генетических аномалий.
Болезнь плотных депозитов (DDD, Dense deposit disease)
Болезнь плотных депозитов – редкая форма патологии, впервые описанная в 1962 г. Galle как новая гломерулярная болезнь, характеризующаяся очень плотной трансформацией ГБМ, выявляемой при ЭМ [39]. В 1975 г. Habib предложил название этой болезни – «болезнь плотных депозитов», рассматривая ее как вариант МПГН II типа [9]. Характерным признаком DDD являются лентовидные отложения очень плотных электронных депозитов в толще ГБМ (интрамембранозно) с трансформацией lamina densa [38, 39] со свечением С3 в мезангиальной области и интрамембранозно [6] при ИФ.
Морфологически данная патология может иметь не только МПГН-паттерн повреждения. В проведенном исследовании Walker et al. показали, что DDD может быть представлена пятью различными вариантами повреждения. Из 67 больных только 25% имели светооптическую картину МПГН. У остальных были обнаружены мезангиопролиферативный ГН (45%), экстракапиллярный ГН с полулуниями (18%), диффузный пролиферативный ГН с экссудативными изменениями (12%); в двух случаях выявленные изменения не удалось классифицировать в рамках какого-либо одного варианта повреждения [20, 38–40].
Как уже упоминалось выше, данное заболевание возникает, как правило, в детском возрасте. Клиническая картина чаще представлена нефротическим синдромом, нередко в сочетании с остронефротическим синдромом, однако возможен и дебют с микрогематурии или субнефротической протеинурии. Течение болезни может быть различным, однако в 50% случаев она прогрессирует до терминальной стадии ХПН в течение 10 лет и в 50% случаев рецидивирует в трансплантате [22, 27, 29, 32, 39, 41], что подтверждает важную роль циркулирующего С3-Неф-фактора в патогенезе [42]. Описана связь между DDD и макулярной дегенерацией сетчатки, а также парциальной липодистрофией [32, 39], которые также относятся к комплемент-опосредованной патологии.
При DDD часто выявляют гипокомплементемию (низкий уровень общей гемолитической активности комплемента CH50) со снижением уровня С3-компонента за счет его безудержного потребления и нормальным уровнем С4, что свидетельствует об активации АПК, а также наличие С3-Неф [21, 43].
Диагностика
В настоящее время МПГН рассматривается как определенный гистологический вариант гломерулярного повреждения с разнообразной этиологией: аутоиммунные заболевания, инфекции, диспротеинемии, дисрегуляция АПК [18]. При исключении инфекции или системного заболевания как причины развившегося почечного повреждения диагностируют первичный, идиопатический, МПГН. Доля идиопатического МПГН в последнее время значительно уменьшилась, поскольку современные диагностические методы позволяют установить этиологию заболевания в большинстве случаев МПГН [18, 44].
Лабораторно у пациентов с МПГН часто отмечается сниженный уровень С3- и/или С4-компонентов комплемента. Снижение обоих показателей характерно для иммунокомплексного нефрита и чаще всего встречается у больных СКВ, т.к. для образования С3-конвертазы КП потребляются и С3, и С4, причем снижение С4 выражено в большей степени, чем С3. Снижение С3 при нормальном уровне С4 зачастую говорит о дисфункции АПК с усиленным потреблением С3 для формирования С3-конвертазы АП, особенно в острую фазу [27]. Однако нормальный уровень С3 не исключает дисфункции АПК [1].
Обновленная классификация разделяет МПГН, основываясь в первую очередь на патогенезе заболевания (рис. 1). Для этого необходимо выполнение всех видов морфологического исследования: световой, электронной и иммунофлюоресцентной микроскопии. Имея только гистологическую картину МПГН при СМ, невозможно поставить нозологический диагноз. Для уточнения типа МПГН необходимо электронно-микроскопическое исследование с выявлением локализации депозитов, а для определения механизма развития нефрита – данные иммунофлюоресценции с информацией о наличии в составе депозитов ИГ и/или компонентов комплемента.
Если полное морфологическое исследование, включающее все 3 вида микроскопии, не выявляет депозитов, следует диагностировать хроническую ТМА даже в отсутствие тромбов [18, 25, 44–46].
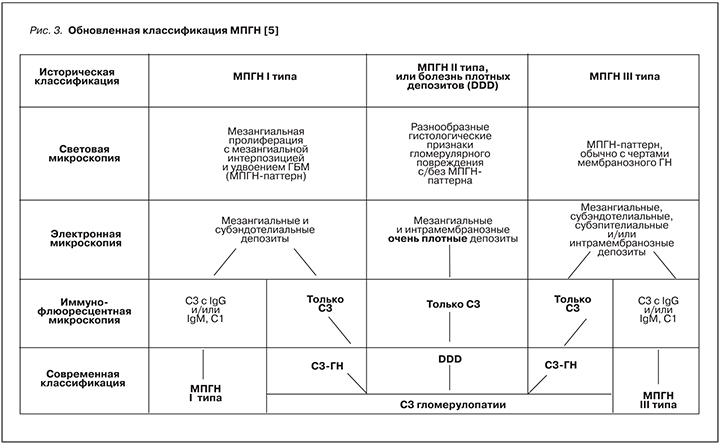
Условно алгоритм диагностики на сегодняшний день можно представить поэтапно (рис. 3). I этап – световая микроскопия, выявляет морфологический вариант повреждения (МПГН). II этап – иммунофлюоресцентная микроскопия, дает представление о составе депозитов (ИГ-позитивный, комплемент-позитивный или ИГ-негативный, комплемент-позитивный МПГН) [4] (рис. 3). В первом случае необходимо исключить вторичный генез нефрита, во втором – можно говорить о наличии С3-гломерулопатии. III этап – электронная микроскопия, выявляет локализацию депозитов, позволяя дифференцировать DDD и С3-ГН, а также различные виды вторичных нозологий.
В связи с новыми данными о патогенезе МПГН, разнообразии этиологических факторов стало очевидно, что при одном и том же морфологическом варианте механизмы развития и прогрессирования заболевания могут кардинально различаться. Это многообразие вариантов МПГН затрудняет лечение и не позволяет сравнивать и оценивать его результаты. Обновленная классификация отражает те новые знания об иммунопатогенезе, которые могут дать толчок поиску новых терапевтических стратегий.
Заключение
Таким образом, сегодня МПГН представляется нам как модель, «паттерн» гломерулярного повреждения, развивающегося при ряде патологий, различающихся этиологией и механизмом развития. Современные представления о патогенезе МПГН позволили выделить отдельную группу заболеваний, имеющих не иммунокомплексный механизм развития, как считалось ранее, а иной – связанный с дисрегуляцией альтернативного пути комплемента. В связи с этим появилась необходимость в создании нового подхода к лечению – антикомплементарной терапии, направленной на подавление спонтанной активации комплемента.


