
Введение
В настоящее время папиллоренальный синдром (ПРС) рассматривается как первичная сосудистая дисгенезия, характеризующаеся поражением зрительного нерва и центральной нервной системы, органов мочевой системы, внутреннего уха [2]. Клинически синдром характеризуется прежде всего патологией глаз (77%) и патологией почек (92%) [3].
Аномалии глаз варьируются от бессимптомных сосудистых аномалий сетчатки до тяжелых нарушений зрительного анализатора и могут включать дисплазию диска зрительного нерва различной степени выраженности, колобому сетчатки, сосудистой оболочки и радужки [4]. Менее распространены сочетанные пороки развития глаз: стафилома склеры, аномалии желтого пятна, аномалии хрусталика, отслоение сетчатки [4]. Острота зрения колеблется от близкой к норме до серьезных нарушений на один или оба глаза (в 75%), также у пациентов может отмечаться нистагм и косоглазие [2].
Проявления со стороны почек включают гипоплазию почек, которая может быть как двусторонней, так и односторонней, мультикистоз почек, пузырно-мочеточниковый рефлюкс (ПМР). Хроническая почечная недостаточность (ХПН) может возникать в любом возрасте, при этом исход в терминальную хроническую почечную недостаточность (тХПН) отмечен почти у всех пациентов.
Примерно у 7% пациентов с ПРС имеет место нарушение слуха в виде высокочастотной сенсоневральной тугоухости; возраст начала потери слуха и его прогрессирования остается неизвестным и не оценивался проспективно [5]. В единичных случаях у пациентов имели место другие аномалии развития: пороки развития нервной и сердечно-сосудистой систем, аномалии развития половых органов, гиперэластичность кожи, слабость связочного аппарата [6].
Многообразие клинических проявлений при ПРС связано с экспрессией гена PAX2 в мезанефросе, эмбриональной щели зрительной чашки, заднем мозге, на ранних стадиях онтогенеза.
Первое упоминание о заболевании было сделано G. Rieger (1977), который сообщил о семье, в которой у отца наблюдались аномалии дисков зрительных нервов и он умер от хронического нефрита. У его сына также имелись аномалии дисков зрительных нервов и сетчатки глаза, при этом функция почек была сохранной; у дочери наблюдались аномалии развития глаз и хроническая почечная недостаточность [7].
Впервые термин ПРС предложен в 1990 г. S. Weaver и соавт. в 1988 г., которые впервые отметили и описали у 2 братьев колобому зрительного нерва и отметили связь этого симптома с заболеванием почек [8]. ПРС наследуется по аутосомно-доминантному типу и возникает в результате мутации одной копии гена PAX2, расположенного на хромосоме 10-го региона q24.3-q25.1 (рис. 1).
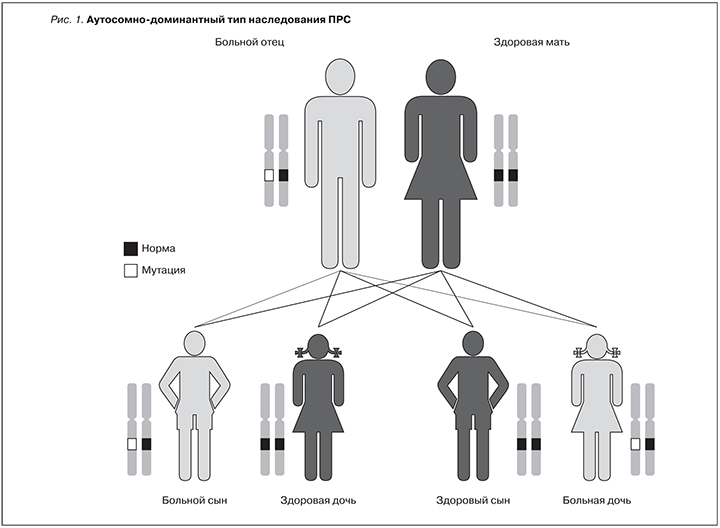
Заболевание обусловлено мутацией в гене PAX2, который кодирует фактор транскрипции парных окон ДНК-связывающих белков, участвующих в развитии урогенитального тракта, зрительного нерва и сетчатки, а также центральной нервной системы и внутреннего уха [9]. Этот ген играет важную роль в эмбриогенезе, запуская молекулярный каскад реакций во время перехода от недифференцированной мезенхимы к мезонефросу [2, 13].
 Кроме того, белок связывает отдельные участки ДНК и регулирует активность других генов [9].
Кроме того, белок связывает отдельные участки ДНК и регулирует активность других генов [9].
Роль мутации в гене PAX2 продемонстрирована в экспериментальных исследовательских работах, проводимых на биологических моделях. У трансгенных мышей была смоделирована мутация в этом гене в тех же областях, что и у пациентов с ПРС. В результате в исследуемой линии мышей обнаружены аномалии развития сосудов сетчатки и дисков зрительных нервов (рис. 2) [9].
При исследовании ткани почек у той же линии мышей продемонстрированы аномалии развития, которые проявлялись в виде двусторонней кистозной дисплазии и гидронефроза. У части мышей имела место односторонняя гипоплазия почки. В редких случаях отмечалось недоразвитие почечных канальцев (рис. 3) [9].
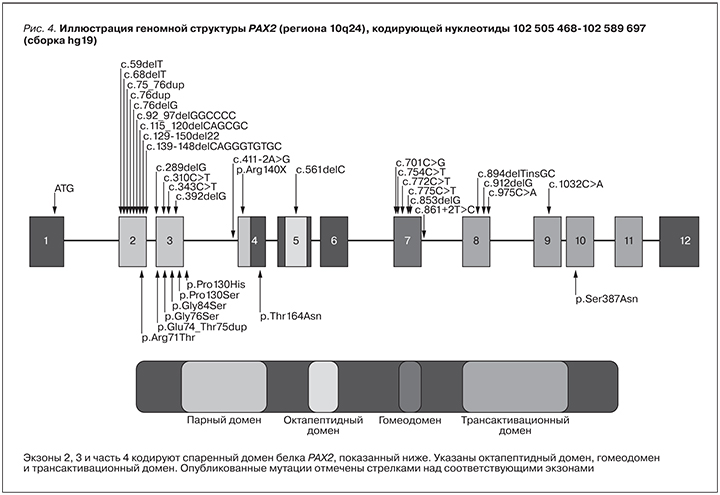
 На сегодняшний день описано более 25 аутосомно-доминантных мутаций при ПРС, идентифицированных в кодирующей области гена PAX2. Большинство этих мутаций встречается в экзонах 2–4 (кодирование парного домена) и в экзонах 7–9 (кодирующих область трансактивации). Наиболее распространенными рекуррентными мутациями являются мутационные изменения в пределах гомогуанинового растяжения (7G) в экзоне 2 (c.76dup, c.76del, c.75_76dup). Эти мутации первоначально были определены как 619insG, 619delG и 619insGG (рис. 4) [9, 10].
На сегодняшний день описано более 25 аутосомно-доминантных мутаций при ПРС, идентифицированных в кодирующей области гена PAX2. Большинство этих мутаций встречается в экзонах 2–4 (кодирование парного домена) и в экзонах 7–9 (кодирующих область трансактивации). Наиболее распространенными рекуррентными мутациями являются мутационные изменения в пределах гомогуанинового растяжения (7G) в экзоне 2 (c.76dup, c.76del, c.75_76dup). Эти мутации первоначально были определены как 619insG, 619delG и 619insGG (рис. 4) [9, 10].
Ряд исследователей отметили, что мутации во 2-м, 3 и 4-м экзонах проявляются преимущественно аномалиями развития глаз без существенных функциональных изменений и патологией почек, тогда как мутации, локализованные в 7-м и 9-м экзонах, связаны с гипоплазией/дисплазией почек [11, 12].
В настоящее время большинство моногенных синдромов рассматривается в рамках врожденных аномалий развития почек и органов мочевой системы, в литературе известных под термином CAKUT (congenital anomalies of the kidney and urinary tract), и являются причиной хронической болезни почек у детей в 40–60% случаев, занимая лидирующее место в этиологии терминальной почечной недостаточности в детской популяции [15]. Для ранней диагностики синдромальных вариантов патологии различные сочетания аномалий развития почек с экстраренальными проявлениями считаем необходимым включать в дифференциальный ряд.
Существует как минимум 20 моногенных вариантов CAKUT (табл), часть из которых наследуется по аутосомно-доминантному типу, обусловливая высокий риск проявления заболевания в последующих поколениях [15, 16]. Большинство семейных вариантов CAKUT приходится на синдромы, обусловленные мутацией генов PAX2 (ПРС), EYA1 (брахиооторенальный синдром), HNF1B (почечные кисты и сахарный диабет).

Нарушение экспрессии мутантных генов в определенный период эмбриогенеза определяет характер поражения мочевой системы, других систем и органов. Известно, что гены BMP4, EYA1, GATA3, PAX2, RET, ROBO2, SALL1, SIX1 и SIX2 имеют высокую экспрессию на ранней стадии эмбриогенеза, когда происходит взаимодействие мочеточникового зародыша и метанефральной мезенхимы. Поэтому мутация данных генов ведет к развитию грубых врожденных пороков органов мочевой системы. В то же время гены AGT, AGTR, UMOD имеют большое значение для формирования нефрона в целом на более поздних стадиях эмбриогенеза [16].
Диагностика ПРС основана на ультразвуковом исследовании (УЗИ) почек, результатах офтальмологического осмотра, семейном анамнезе и молекулярно-генетическом тестировании. С учетом международных рекомендаций при подозрении на ПРС ребенку необходимо комплексное обследование мочевой системы, органов зрения и консультация специалистов: нефролога, уролога, офтальмолога, сурдолога, детского гинеколога, невропатолога, клинического генетика.
Лечение заболевания направлено на предотвращение осложнений почечной недостаточности и потери зрения в результате отслойки сетчатки.
Как и для большинства пациентов с хронической болезнью почек, показано раннее назначение ренопротективной терапии ингибиторами ангиотензинпревращающего фермента [15, 17]. Лечение ХПН в терминальной стадии требует своевременного проведения заместительной почечной терапии (диализа и трансплантации почек).
Пренатальная диагностика включает УЗИ плода беременной женщины. Считают, что оптимальное время для проведения молекулярно-генетического тестирования у беременных группы риска в 15–18 недель гестации. Проводят анализ ДНК, извлеченного из эмбриональных клеток, полученных путем амниоцинтеза или биопсии хориона.
Ниже приведено клиническое наблюдение за ребенком с ПРС, которое представляет интерес с точки зрения клинического течения и ведения ребенка с данной патологией.
Клинический случай
Больной 2007 г. рождения обследован в отделе наследственных и приобретенных болезней почек НИКИ педиатрии им. акад. Ю.Е. Вельтищева РНИМУ им. Н.И. Пирогова. При обследовании ребенка применялись клинико-генеалогический метод, функциональные методы исследования (УЗИ почек и мочевого пузыря, внутривенная урография и цистография, УЗИ глаз, МРТ головного мозга, денситометрия), клиническое и биохимическое исследование крови и мочи.
В ООО «Генотек» ребенку проведено секвенирование экзома человека (44М) путем селекции специфичных фрагментов ДНК с помощью системы SureSelect с последующим параллельным секвенированием полученных библиотек по технологии Illumina. Мутация подтверждена методом по Сенгеру на секвенаторе Applied Biosystems 3500 в НИЛ общей патологии института.
Ребенок Д. 2007 г. рождения поступил в отделение нефрологии НИКИ педиатрии им. акад. Ю.Е. Вельтищева РНИМУ им. Н.И. Пирогова в возрасте 5 лет с жалобами на изменения почек (двусторонняя гипоплазия, нефрокальциноз) по данным УЗИ.
Мальчик Д. из семьи с отягощенной наследственностью. У отца ребенка в возрасте 18 лет выявлена тХПН, причина которой неизвестна, он перенес 2 трансплантации почек в 19 и в 25 лет. Мальчик от 1-й беременности, протекавшей на фоне анемии, роды в срок на 38-й неделе гестации с нормальными массоростовыми показателями. Физическое и нервно-психическое развитие соответствует возрасту. С 2 месяцев жизни у ребенка выявлены абактериальная лейкоцитурия, минимальная протеинурия, непостоянная глюкозурия, а также уменьшение возрастных размеров почек по данным УЗИ. Наблюдался по месту жительства с диагнозом «двусторонняя гипоплазия почек, вторичный пиелонефрит». При поступлении в отделение нефрологии института в возрасте 5 лет физическое развитие среднее гармоничное. При осмотре обращало на себя внимание наличие микроаномалий развития: гипертелоризм сосков, брахидактилия стоп и кистей, широкая сандалевидная щель, широкие дистальные фаланги пальцев, приросшие ушные мочки. При обследовании мочевой синдром был представлен в виде непостоянной глюкозурии, высоким уровнем β2-микроглобулина мочи.
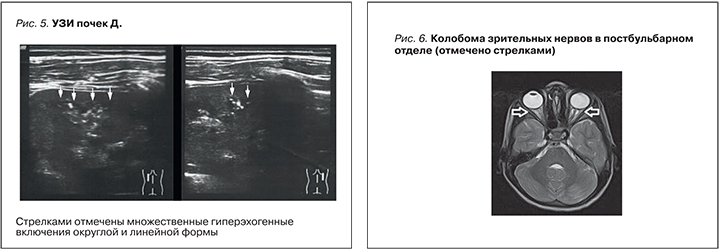
В биохимическом анализе крови отмечено повышение уровня мочевины до 11,4 ммоль/л, снижение расчетной скорости клубочковой фильтрации (рСКФ), рассчитанной по формуле Шварца, до 77 мл/мин/1,73 м2. Нарушений обмена фосфора и кальция не выявлено. Уровень паратгормона и гормонов щитовидной железы в пределах референcных значений. Кислотно-основное состояние крови и электролиты в пределах нормальных значений. По данным биохимического анализа мочи, экскреция солей без особенностей. При исследовании аминокислотного спектра мочи выявлена неселективная аминоацидурия. По данным УЗИ, размеры почек уменьшены и соответствуют 5‰, эхогенность паренхимы повышена, сравнима с эхогенностью печени, плохо дифференцирована, множественные гиперэхогенные включения округлой и линейной форм размером до 0,25 см (рис. 5). При экскреторной урографии рентген-контрастных включений не выявлено. Отмечается уменьшение левой почки на 32% по площади, правой – на 20%. Функция почек по визуальной оценке не нарушена.
При проведении микционной цистографии данных за ПМР нет. Мальчик осмотрен офтальмологом, проведено УЗИ глаз, где выявлена аномалия развития дисков зрительных нервов.
В связи с этим была проведена магнитно-резонансная томография (МРТ) головного мозга, которая выявила колобому зрительных нервов в постбульбарном отделе (рис. 6).
Ребенок осмотрен сурдологом – патология слуха не выявлена. По данным рентгенограммы костей голени, форма и структура костей не изменены, индекс Бернарда Лаваля – 0,38. Проведена денситометрия для выявления признаков локального остеопороза, где данных за общий остеопороз не выявлено.
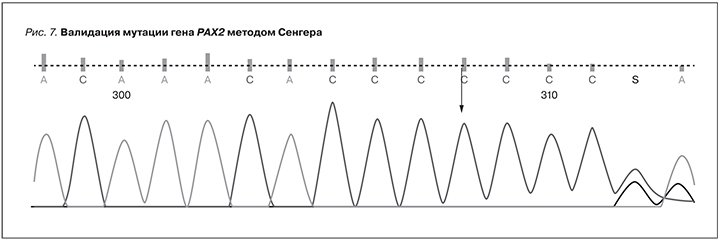
С учетом данных анамнеза и обследования консультирован генетиком и заподозрен ПРС, в связи с чем проведено молекулярно-генетическое исследование, по данным которого во 2-м экзоне гена PAX2 выявлена ранее описанная в литературе у больных ПРС гетерозиготная инсерция гуанина в 76-м положении, приведшая к сдвигу рамки считывания, начиная с аминокислоты валин в положении 25 в молекуле белка. Выявленная мутация валидирована по методу Сенгера (рис. 7).
Учитывая отягощенную наследственность, комплекс микроаномалий развития, гипоплазию почек, нефрокальциноз, снижение рСКФ, признаки проксимальной тубулярной дисфункции, колобому дисков зрительных нервов и результаты молекулярно-генетического исследования выставлен диагноз «ПРС. Хроническая болезнь почек 2-й стадии». Осложнение: медулярный нефрокальциноз 2-й степени.
Ребенку проведена терапия: повышенный питьевой режим, ингибиторы кристаллообразования (блемарен) постоянно под контролем кислотности мочи.
В динамике наблюдения за ребенком отмечена нормализация уровня мочевины крови, повышение уровня рСКФ до 89 мл/мин/1,73 м2. Также наблюдалась положительная динамика по УЗИ-картине почек в виде уменьшения размеров кальцинатов с 0,25 до 0,10 см. В настоящее время мальчик продолжает наблюдаться в отделении нефрологии института, продолжает получать терапию.
Обсуждение
Представленное клиническое наблюдение подтверждает, что ядром клинической картины ПРС является совокупность клинических признаков: двусторонняя гипоплазия почек и колобома зрительных нервов. Клинические наблюдения ПРС уже описаны в литературе, однако особенностью данного клинического примера служит наличие признаков проксимальной канальцевой дисфункции (непостоянная глюкозурия, неселективная аминоацидурия, высокий уровень β2-микроглобулина мочи), которые, по данным литературы, не встречались у описанных ранее пациентов. Наличие у пациента проксимальной тубулярной дисфункции на первоначальном этапе обследования заставило проводить дополнительный дифференциально-диагностический поиск для исключения болезней, связанных с патологией обмена кальция и фосфора, нарушением кислотно-основного состояния и др. А сочетание аномалий развития зрительных нервов и почек заставило исключить ряд моногенных синдромов с поражением органов зрения и почек: синдромы COACH (Cerebellar vermis hypoplasia, Oligophrenia, congenital Ataxia, ocular Coloboma, Hepatic fibrosis), Жубера, CHARGE и др. По данным литературы, в подавляющем большинстве случаев средний возраст развития тХПН при ПРС составляет 19 лет, что, вероятно, наблюдалось и у отца ребенка. Снижение рСКФ у нашего пациента уже в дошкольном возрасте определяет неблагоприятный прогноз заболевания в плане развития тХПН, а наличие проксимальной тубулярной дисфункции свидетельствует о тяжести поражения не только гломерул, но и тубулярного отдела почек.
Таким образом, на примере редкого моногенного синдрома показана необходимость комплексного подхода к обследованию детей с аномалией развития почек для ранней диагностики синдромальных вариантов патологии, что важно для определения нефрологического, витального, социального и семейного прогнозов заболевания.


