Введение
Хроническая почечная недостаточность (ХПН) служит исходом многих заболеваний, сопровождающихся повреждением почек. Терминальная ХПН ассоциирована с высокой смертностью и необходимостью применения дорогостоящих методов заместительной почечной терапии (ЗПТ) – диализа и пересадки почки [1].
Прогноз пациентов с поздними стадиями заболевания наряду со скоростью снижения функции почек и эффективностью ЗПТ определяется наличием осложнений: гипертензии, анемии, тромбозов, гипертрофии левого желудочка, наличием и выраженностью минерально-костных нарушений. Наиболее ранним и частым спутником хронической болезни почек (ХБП) является анемия, которая вносит существенный вклад в прогрессирование кардиоваскулярных нарушений. В больших обсервационных исследованиях показано, что у пациентов с ХБП 3–5-й ст. снижение уровня гемоглобина ассоциировано с повышением риска смерти и сердечно-сосудистых осложнений, а также с существенным снижением качества жизни, обусловленным выраженной слабостью, одышкой, бессонницей, головной болью, снижением умственной и физической активностей [2]. Закономерно, что в большинстве случаев частота и тяжесть анемии коррелируют со снижением скорости клубочковой фильтрации (СКФ). По данным McClellan et al., доля пациентов с анемией среди больных с СКФ ≥60 мл/мин/1,73 м2 составляет 26,7%, тогда как в когорте пациентов с СКФ <15 мл/мин/1,73 м2 уровень гемоглобина ≤120 г/л регистрируется в 75,5% случаев [2].
Ввиду того что основной причиной развития нефрогенной анемии является дефицит эндогенного эритропоэтина (ЭПО) и функциональный дефицит железа, данное состояние потенциально обратимо. ЭПО-терапию для коррекции анемии получают примерно 20% пациентов с ХБП 4-й ст. и 42% пациентов с ХБП 5-й ст. до диализа [3] и около 70% пациентов на гемодиализе (ХБП-5Д).
Оптимальные подходы к лечению анемии почечного генеза продолжают обсуждаться и пересматриваться. На разных стадиях клинических исследований находятся противоанемические препараты для перорального применения новых классов, в т.ч. стабилизаторы фактора, индуцированного гипоксией, – ингибиторы пролил гидралазы. Однако до настоящего времени стандартом терапии остается использование препаратов железа и эритропоэзстимулирующих лекарственных средств (ЭСС). Согласно рекомендациям NICE, кандидатами для проведения противоанемической терапии ЭСС являются все пациенты, у которых данная тактика может приводить к достижению целевых значений уровня гемоглобина (10–12 г/дл), улучшению качества жизни, повышению физической активности и отказу от гемотрансфузий [4].
К ЭСС относятся препараты рекомбинантного человеческого эритропоэтина альфа и бета короткого действия (рчЭПО), дарбэпоэтин альфа и метоксиполиэтиленгликоль – эпоэтин бета. Результаты опубликованных в настоящее время исследований не позволяют говорить об однозначном превосходстве одних ЭСС над другими. По мнению экспертов, выбор конкретного ЭСС для коррекции анемии должен быть индивидуализирован и определяться балансом фармакокинетических и фармакодинамических характеристик препарата, данных о его безопасности, стоимости, доступности, удобстве применения в конкретной ситуации и предполагаемой пользы для больного, в частности повышения качества жизни [5].
Несомненным преимуществом применения любых ЭСС для коррекции нефрогенной анемии являются в большинстве случаев возможность полного отказа от гемотрансфузий и контроль симптомов анемии. В то же время существуют данные о повышении риска смерти и сердечно-сосудистых осложнений (инсульта, тромбоза сосудистого доступа, гипертензии) на фоне агрессивной противоанемической терапии. Однако, по мнению ряда авторов, развитие указанных нежелательных явлений в большей степени связано с превышением целевого уровня гемоглобина, высокой вариабельностью данного показателя, чем с фактом применения ЭСС [6]. Следовательно, одной из наиболее важных задач при применении ЭСС является поддержание уровня гемоглобина в пределах рекомендованных целевых значений и минимизация его флуктуаций. Таким образом, своевременная коррекция дозы ЭСС представляется одним из ключевых условий безопасного лечения анемии. Осуществлять изменение дозы препарата целесообразно сразу после достижения целевого уровня гемоглобина, не дожидаясь повышения его уровня >120 г/л. При этом в идеале эффект коррекции должен начинаться уже через 2 недели после изменения дозы, что соответствует срокам развития зрелых эритроцитов от стадии коммитированных клеток – предшественников эритропоэза [7]. С этой точки зрения наиболее приемлемыми представляются рчЭПО короткого действия, обладающие коротким периодом полувыведения, инъекции которых осуществляются 3 раза в неделю. Однако, принимая во внимание такие аспекты, как флуктуации гемоглобина, комплаентность, удобство более редкого введения, препаратами выбора становятся пролонгированные ЭСС, к примеру дарбэпоэтин альфа.
Дарбэпоэтин альфа имеет молекулярную массу 37,1 кДа, 5 участков гликозилирования, 22 свободные сиаловые группы. Вследствие повышенного содержания углеводов дарбэпоэтин альфа обладает более длительным периодом полувыведения по сравнению с рчЭПО, а следовательно, и большей активностью in vivo. В рамках клинических исследований показано, что дарбэпоэтин альфа, вводимый 1 раз в неделю или 1 раз в 2 недели, столь же эффективен, как и рчЭПО короткого действия при стандартных режимах терапии [8, 19]. Таким образом, применение дарбэпоэтина альфа позволяет сохранить баланс между эффективностью лечения, возможностью своевременной коррекции дозы препарата для обеспечения безопасности терапии и удобством применения.
В РФ применение оригинального препарата дарбэпоэтина альфа Аранесп (Амджен Европа Б.В., Нидерланды) ограничено его высокой стоимостью. Согласно общемировой тенденции разработки и внедрения в практику аналогов дорогостоящих биологических препаратов с целью повышения доступности терапии, компанией ЗАО «БИОКАД» был разработан препарат Дарбэстим (дарбэпоэтин альфа), представляющий собой биоаналог препарата Аранесп.
В процессе создания и последующего изучения препарата Дарбэстим соблюдены все международные требования для разработки биоаналогов препаратов эритропоэтина [9]. На первом этапе проведены обширные доклинические исследования, в ходе которых установлена эквивалентность физико-химических и биологических свойств препаратов Дарбэстим и Аранесп (табл. 1).

Результаты проведенных доклинических исследований позволили приступить к клиническим исследованиям с участием человека (клинические исследования I и III фаз).
Проведено два клинических исследования I фазы с участием здоровых добровольцев. В первом исследовании BCD-066-1 проведено изучение и сравнение фармакокинетики, фармакодинамики, безопасности и иммуногенности препаратов Дарбэстим и Аранесп при их однократном подкожном и однократном внутривенном введениях. Во втором исследовании I фазы, BCD-066-3, проведено сравнение фармакокинетики, фармакодинамики, безопасности и иммуногенности препаратов при их многократном внутривенном введении. Результаты обоих исследований показали, что по своим фармакокинетическим и фармакодинамическим свойствам, безопасности и иммуногенности исследуемый препарат Дарбэстим эквивалентен препарату Аранесп.
Согласно рекомендациям Европейского медицинского агентства (European Medicines Agency, EMA), заключительным этапом разработки биоаналогов препаратов, содержащих эритропоэтин, должно стать проведение клинического исследования III фазы в популяции пациентов с дефицитом эндогенного эритропоэтина и с сохраненной чувствительностью костного мозга к воздействию эритропоэтина [9]. Клиническое исследование III фазы BCD-066-2 «Многоцентровое двойное слепое рандомизированное сравнительное клиническое исследование в параллельных группах эффективности и безопасности препаратов BCD-066 (Дарбэстим) (ЗАО «БИОКАД», Россия) и Аранесп (Амджен Европа Б.В., Нидерланды) в терапии анемии у больных хронической почечной недостаточностью, находящихся на гемодиализе» проведено с целью доказательства эквивалентной эффективности и безопасности препаратов Дарбэстим и Аранесп. В основе данного клинического исследования лежала гипотеза, согласно которой клиническая эффективность препарата BCD-066 (Дарбэстим), оцененная на основании сравнения изменения уровня гемоглобина от начала исследования и до периода оценки эффективности, эквивалентна эффективности препарата Аранесп. Первичный анализ эффективности, безопасности и иммуногенности исследуемых препаратов проведен через 24 недели терапии, его результаты представлены в настоящей статье.
Материал и методы
Исследование проведено в соответствии с этическими принципами, изложенными в Хельсинкской декларации Всемирной медицинской ассоциации, требованиями Федерального законодательства (№ 61-ФЗ от 12.04.2010) и правилами Национального стандарта РФ «Надлежащая клиническая практика» (ГОСТ Р 52379-2005), ICH GCP и другими нормативными требованиями [10, 11, 12, 13]. В общей сложности набор и лечение пациентов осуществлены на базе 23 исследовательских центров Российской Федерации. Все документы клинического исследования рассмотрены и одобрены Советом по этике и локальными этическими комитетами всех исследовательских центров.
Дизайн исследования: рандомизированное двойное слепое сравнительное клиническое исследование в параллельных группах.
Критерии включения в исследование: наличие письменного информированного согласия, возраст от 18 до 75 лет, установленный диагноз терминальной стадии почечной недостаточности, потребность в проведении гемодиализа общей продолжительностью не менее 12 часов в неделю в течение как минимум последних 3 месяцев до даты рандомизации, регулярное введение рчЭПО в стабильной дозе и со стабильной частотой (1, 2 или 3 раза в неделю) в течение не менее 3 месяцев до даты рандомизации, целевой уровень гемоглобина (100–120 г/л) в течение 3 месяцев до даты рандомизации, установленная эффективность гемодиализа (индекс диализной дозы [Kt/v] ≥1,2), коэффициент насыщения трансферрина ≥20%, уровень ферритина >100 нг/мл. В исследование не включались пациенты с любыми иными формами анемии, за исключением почечной, установленным диагнозом волчаночного нефрита или наличием хронического заболевания почек, развившегося как следствие системного васкулита.
До начала любых процедур на скрининге пациенты подписывали информированное согласие. Информация для пациента содержала все сведения о настоящем клиническом исследовании, необходимые для принятия осмысленного и самостоятельного решения.
В скрининговом периоде на основании данных анамнеза и клинико-лабораторного обследования произведена оценка соответствия пациентов критериям включения/невключения. В рамках данного исследования осуществлена стратификация пациентов по наличию/отсутствию хотя бы одного из факторов риска сосудистых тромботических осложнений (возраст ≥60 лет, наличие сосудистых протезов, наличие сахарного диабета), что позволило добиться того, чтобы группы были равнозначными по количеству пациентов с вышеперечисленными признаками.
Участники исследования были рандомизированы в две группы терапии в соотношении 1:1. Пациенты группы 1 (n=98) получали исследуемый препарат дарбэпоэтина альфа Дарбэстим, пациенты группы 2 (n=98) – референтный препарат Аранесп. Введение обоих препаратов осуществлено подкожно, еженедельно, после завершения сеанса гемодиализа в середине недели – в общей сложности в течение 52 недель. Исходная еженедельная доза препарата рассчитывалась в зависимости от дозы рчЭПО, которую пациент получал до включения в исследование, по формуле: доза дарбэпоэтина альфа (мкг/нед)=общая еженедельная доза рчЭПО (МЕ/нед)/200. В случае если до включения в исследование пациент получал дарбэпоэтин альфа, доза его оставалась прежней. Коррекция вводимой дозы в дальнейшем осуществлялась с учетом уровня гемоглобина. В течение всего исследования препараты были заслеплены.
Исследование включило два этапа. Первый (основной) продолжался с 1-й по 24-ю недели терапии включительно. После его окончания выполнен анализ эффективности, безопасности и иммуногенности препаратов, результаты которого представлены в данной статье. Дополнительный этап исследования продолжался с 25-й по 52-ю неделю исследования включительно (в течение дополнительных 28 недель). С целью мониторинга безопасности после завершения 52-недельного заслепленного периода применения исследуемых препаратов дарбэпоэтина альфа и назначения последующей терапии за всеми пациентами продолжалось наблюдение на протяжении 4 недель.
Основной анализ эффективности проведен в популяции пациентов, прошедших все предусмотренные протоколом клинического исследования визиты основного 24-недельного этапа исследования (n=176), рer-рrotocol-анализ.
Первичной конечной точкой оценки эффективности стало изменение уровня гемоглобина в течение периода оценки (среднее арифметическое минимум двух определений уровня гемоглобина в период с 21-й по 24-ю неделю исследования) по сравнению с исходным уровнем гемоглобина (среднее арифметическое между уровнем гемоглобина на скрининге и на визите 1 до первого введения исследуемых препаратов).
Основные вторичные конечные точки оценки эффективности: доля пациентов с целевым уровнем гемоглобина (100–120 г/л) в период с 21-й по 24-ю неделю исследования, средняя доза исследуемого препарата (мкг) в течение последних 4 недель лечения в рамках основного периода (неделя 21–неделя 24), доля пациентов, которым потребовалось проведение гемотрансфузий в течение всего основного периода лечения (недели 1–неделя 24). Оценивались также средний уровень гемоглобина в течение всего 24-недельного периода исследования (неделя 1–неделя 24) и в течение последних 4 недель лечения в рамках основного этапа (недели 21– неделя 24), динамика уровня гемоглобина в течение всего основного периода участия пациента в основной части исследования (неделя 1–неделя 24).
Таким образом, для первичной конечной точки и для части вторичных конечных точек в рамках основного периода оценка эффективности производилась с 21-й по 24-ю неделю исследования, для части вторичных конечных точек – с 1-й по 24-ю неделю исследования.
Оценка безопасности осуществлена в популяции пациентов, получавших хотя бы одно введение исследуемого препарата или препарата сравнения, mITT-анализ (n=195) и, согласно конечным точкам оценки безопасности, включала оценку частоты нежелательных явлений (НЯ) и серьезных нежелательных явлений (СНЯ), в.ч. НЯ 3–4-й степеней тяжести, определение частоты случаев досрочного прекращения участия в исследовании из-за развития НЯ или СНЯ, анализ частоты артериальных и венозных тромботических осложнений (нарушение мозгового кровообращения, тромбоз артериовенозного доступа и т.п.). Степень тяжести нежелательных явлений и лабораторных отклонений оценивалась в соответствии с классификацией СТСАЕ v.4.03 (Common Terminology Criteria for Adverse Events) [14]. Поскольку в исследовании принимали участие пациенты с ХПН, получавшие гемодиализ, которые в большинстве случаев имеют сопутствующие заболевания, в целом вероятность развития НЯ и СНЯ в данной популяции была высокой.
В связи с этим особый интерес представлял анализ частоты НЯ и СНЯ, связанных с исследуемой терапией. Оценка связи НЯ и СНЯ с терапией проводилась исследователем. НЯ считалось связанным с исследуемым препаратом, если существовала хотя бы минимальная возможность наличия причинно-следственной связи с ним, т.е. степень связи расценивалась исследователем как «определенная», «вероятная» и «возможная». В остальных случаях (связь «сомнительная», «условная» или «несвязанная») НЯ считалось не связанным с исследуемой терапией.
Согласно протоколу клинического исследования, конечными точками для оценки иммуногенности служили доля пациентов с выявленными связывающими антителами (САТ) и доля пациентов с выявленными нейтрализующими антителами (НАТ) к дарбэпоэтину альфа. В анализ иммуногенности включены все пациенты (n=192), получившие хотя бы одно введение исследуемого препарата или препарата сравнения и имевших результаты исследования на скрининге и хотя бы на одном из последующих визитов. Определение наличия САТ против дарбэпоэтина альфа в сыворотке крови пациентов проведено с использованием валидированных методов твердофазного иммуноферментного анализа с применением пероксидазы хрена в качестве индикаторного фермента в линейном диапазоне концентраций связывающих антител против дарбэпоэтина альфа от 31,25 до 2000 нг/мл. Вначале проводился анализ на наличие САТ (скрининговый и подтверждающий анализы), в случае их обнаружения предполагалось проведение исследования на наличие НАТ.
Расчет выборки
Для определения размера выборки, необходимого для получения достоверных результатов сравнения препаратов Дарбэстим и Аранесп по основной конечной точке эффективности, использованы данные двух рандомизированных клинических исследований референтного препарата Аранесп (NESP980117 и NESP980200): рассчитана объединенная выборочная дисперсия для среднего изменения уровня гемоглобина у пациентов, получавших дарбэпоэтин альфа, и величина различия данного показателя эффективности [15]. В качестве клинически незначимой разницы уровня гемоглобина в достижении ответа между оригинальным препаратом дарбэпоэтина и его биоаналогом принято рекомендованное руководством ЕМА значение, равное 0,5 г/дл (5 г/л) [9]. Расчет популяции исследования произведен по формуле определения размера выборки для гипотезы эквивалентности в параллельных группах при условии равной рандомизации [16, 17]. В расчете использовались следующие значения: ошибка первого рода α=0,05, предполагаемая мощность исследования должна составлять не менее 80%, т.е. ошибка второго рода β=0,2.
Таким образом, при условии участия в исследовании двух групп пациентов и рандомизации в соотношении 1:1 включение 196 человек с учетом возможного 25%-ного досрочного выбывания было достаточным для получения статистически достоверных различий между препаратами или доказательства их отсутствия.
Рандомизация
Проведена центральная рандомизация пациентов в 2 группы в соотношении 1:1 с применением метода блочной рандомизации. С целью обеспечения максимальной равноценности групп перед проведением рандомизации осуществлена стратификация пациентов по критерию наличия/отсутствия хотя бы одного из факторов риска сосудистых тромботических осложнений (возраст ≥60 лет, наличие сосудистых протезов, наличие сахарного диабета). Участников исследования рандомизировали внутри каждой страты. Исследователю сообщался только идентификационный номер пациента и номер лота препарата, назначенного участнику.
Статистический анализ произведен с применением языка программирования для статистической обработки данных R, пакета статистических программ Statistica 10.0 (StatSoft, USA).

Основные демографические и исходные клинико-лабораторные показатели, полученные на скрининге, обрабатывались по правилам описательной статистики и анализировались в популяциях для анализа эффективности и безопасности.
Выбор метода описательной статистики и статистического сравнения определялся типом данных и видом распределения. Для описания количественных переменных, распределенных по нормальному закону (среднее значение изменения уровня гемоглобина в течение основного периода исследования), использовались средние значения и стандартные отклонения; для статистической обработки использовались двухвыборочный критерий Стьюдента и дисперсионный анализ. Для количественных данных, распределенных по отличному от нормального закону распределения (исходные физиологические и клинико-лабораторные характеристики, средний уровень гемоглобина, зарегистрированный с 21-й по 24-ю неделю терапии, средняя доза дарбэпоэтина альфа, вводимая пациентам с 21-й по 24-ю неделю), средние значения описывались с помощью медиан и интерквартильных размахов; для статистической обработки применялись критерии Манна–Уитни и Вилкоксона.
Для описания категориальных данных использованы проценты или доли. Статистическое сравнение категориальных данных проведено с использованием точного теста Фишера или критерия χ2 Пирсона.
В итоговый статистический анализ были включены следующие пациенты:
- Анализ безопасности: все больные, получавшие хотя бы одно введение исследуемого препарата или препарата сравнения (n=195);
- Анализ эффективности: все пациенты, прошедшие все визиты основного этапа исследования (24 недели) (популяция рer-рrotocol, n=176).
Проверка статистической гипотезы эквивалентности с использованием первичной конечной точки (изменение уровня гемоглобина) произведена путем сравнения границ 95% доверительного интервала (ДИ) разности между средними арифметическими значениями изменения уровня гемоглобина в группах с интервалом [-5,00; 5,00], где 5,00 г/л (0,5 г/дл) – граница эквивалентности. Препараты признавались эквивалентными, если границы 95% ДИ для разности средних значений колебаний уровня гемоглобина не выходили за пределы указанной границы эквивалентности. Изменение уровня гемоглобина у каждого из пациентов в обеих группах рассчитывалось как разность между средним значением уровня гемоглобина в течение последних 4 недель лечения (определяемым как среднее арифметическое всех значений уровня гемоглобина [не менее двух], определенных в течение недель 21–24 основного периода исследования) и средним исходным значением уровня гемоглобина (определяемым как среднее арифметическое уровня гемоглобина на скрининговом визите и визите 1 до первого введения).
Результаты
Основной период исследования продолжался с 07.08.2015 (дата первого стартового визита) по 08.06.2017 (дата завершения 24 недель исследования последним участником).
В общей сложности в исследование были включены 196 пациентов с анемией, обусловленной терминальной ХПН. До включения в исследование абсолютное большинство (93,37%, n=183) больных получали рчЭПО короткого действия, 6,63% (n=13) получали ЭСС длительного действия. Участники исследования были рандомизированы в соотношении 1:1 в две группы терапии: 98 пациентов вошли в первую группу и получали препарат Дарбэстим; 98 пациентов были включены в группу 2 и получали Аранесп. Один пациент, включенный во вторую группу, выбыл из исследования до первого введения препарата в связи с развитием СНЯ. Таким образом, 195 пациентов получили хотя бы одно введение исследуемого препарата или препарата сравнения: 98 пациентов первой группы и 97 пациентов второй. Все они были включены в анализ безопасности (популяция mITT).
В течение первых 24 недель терапии из исследования выбыли 20 пациентов. Из группы 1 (Дарбэстим) выбыли 12 человек: 5 пациентов по причине развития НЯ/СНЯ (связь с препаратом не исключалась только в 1 случае), 3 пациента в связи с отзывом информированного согласия, 2 – в связи с нарушением критериев включения/невключения, 2 – в связи с трансплантацией почки. Из группы 2 (Аранесп) в указанный период выбыли 8 человек: 1 пациент по причине развития НЯ/СНЯ (связь с препаратом не исключалась), 2 пациента в связи с нарушением критериев включения/невключения, 2 в связи со сменой диализного центра, 2 в связи с трансплантацией почки, 1 пациент выбыл до первого введения препарата в связи с развитием СНЯ. Таким образом, завершили участие в основном 24-недельном этапе исследования 176 пациентов (86 пациентов в группе 1 и 90 пациентов в группе 2). Данная когорта составила популяцию для основного анализа эффективности – популяция per-protocol (n=176). Распределение пациентов по группам исследования представлено на рис. 1.
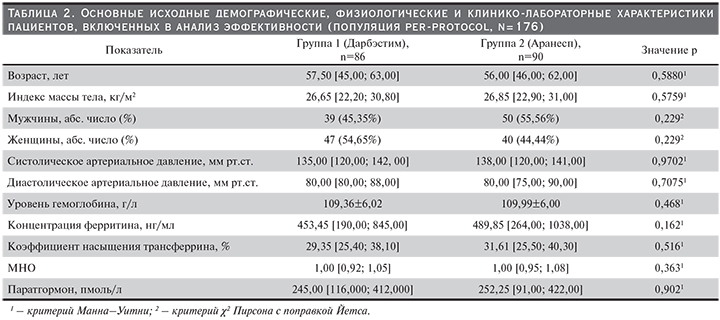
Группы 1 (Дарбэстим, n=86) и 2 (Аранесп, n=90) были сопоставимы по основным исходным демографическим, физиологическим и клинико-лабораторным показателям, исследованным на скрининге. Статистический анализ не выявил достоверных различий между группами (табл. 2).
Оценка эффективности
Первичная конечная точка
В первой группе (Дарбэстим, n=86) среднее значение изменения уровня гемоглобина составило 4,7±11,0 г/л, во второй (Аранесп, n=90) – 4,6±9,0 г/л. Сравнение полученных величин изменения уровня гемоглобина не выявило статистически значимых различий между группами (p=0,9717, t-критерий Стьюдента), табл. 3.
Для подтверждения основной гипотезы исследования об эквивалентной эффективности препаратов Дарбэстим и Аранесп рассчитан классический 95% ДИ для разницы средних арифметических значений показателя «изменение уровня гемоглобина в течение периода оценки по сравнению с исходным уровнем» между группами, который составил [-3,04; 2,93]. Согласно руководству EMA по исследованию воспроизведенных препаратов, содержащих рекомбинантный эритропоэтин, граница эквивалентности эффективности составляет 5,00 г/л, и попадание ДИ внутрь предустановленных границ [-5,00; 5,00] свидетельствует об эквивалентной эффективности двух сравниваемых препаратов [9]. Таким образом, доказана эквивалентная эффективность препаратов Дарбэстим и Аранесп, первичная конечная точка исследования достигнута.
Вторичные конечные точки
С 21-й по 24-ю неделю терапии уровень гемоглобина находился в пределах целевых значений (100–120 г/л) и ни разу за этот период не выходил за их границы у 70,9% (n=61) пациентов, получавших Дарбэстим, и у 67,8% (n=61), получавших Аранесп. Статистически значимых различий по данному показателю между группами не выявлено (р=0,772, критерий χ2 Пирсона с поправкой Йетса).
Средняя доза дарбэпоэтина альфа, вводившаяся пациентам с 21-й по 24-ю неделю терапии, была одинаковой в первой и второй группах и составила 20 [10; 30] мкг (p=0,8165, критерий Манна–Уитни).
На протяжении 24 недель основного этапа исследования ни одному пациенту ни в первой, ни во второй группах не потребовалось проведения гемотрансфузии.
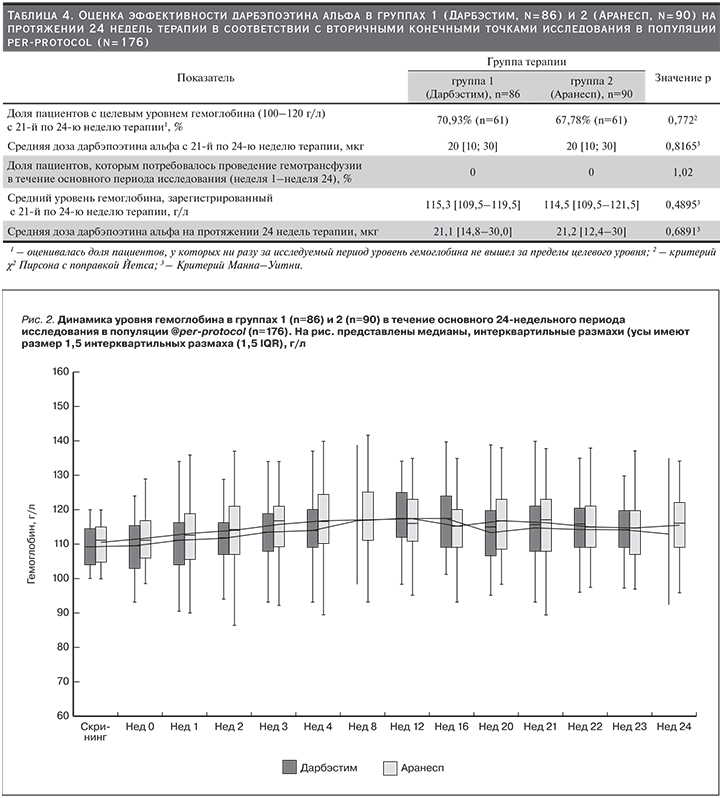
Еще одним критерием оценки эффективности препаратов служил средний уровень гемоглобина, зарегистрированный с 21-й по 24-ю неделю терапии. В группах 1 (Дарбэстим) и 2 (Аранесп) данный показатель составил 114,5 [109,5–121,5] и 115,3 [109,5–119,5] г/л соответственно (р=0,4895, критерий Манна–Уитни). Таким образом, в указанный период средний уровень гемоглобина в группах статистически значимо не различался и находился в пределах «целевых» значений. Важно отметить, что при анализе среднего уровня гемоглобина на каждом из визитов на протяжении всех 24 недель терапии как в первой, так и во второй группе данный показатель также находился в пределах «целевых» значений 100–120 г/л (рис. 2).
На протяжении 24 недель терапии средняя доза дарбэпоэтина альфа составила 21,1 [14,8–30,0] и 21,2 [12,4–30] мкг в группах 1 и 2 соответственно (р=0,6891, критерий Манна–Уитни). Отсутствие статистически значимых различий между двумя группами по данному показателю свидетельствует в пользу эквивалентной эффективности исследуемого препарата и препарата сравнения.
Таким образом, полученные результаты свидетельствуют о том, что исследуемый препарат Дарбэстим и препарат сравнения Аранесп были одинаково эффективными для поддержания «целевых» значений уровня гемоглобина при использовании аналогичных доз дарбэпоэтина альфа (мкг).
В табл. 4 представлены результаты оценки эффективности препаратов дарбэпоэтина альфа в группах 1 (n=86) и 2 (n=90) в соответствии с вторичными конечными точками исследования.
Оценка безопасности
В анализ безопасности включены 195 пациентов (популяция mITT): 98 – первой группы, получавших препарат Дарбэстим, и 97 – второй, получавших препарат Аранесп. Абсолютное большинство участников исследования имели одно или более сопутствующее заболевание. Число таких пациентов, а также спектр нозологий были сопоставимыми в обеих группах. Превалировали сердечно-сосудистые заболевания: артериальная гипертензия наблюдалась у 61,22% (n=60) пациентов первой группы и у 55,67% (n=54) – второй (р=0,5211, критерий χ2 Пирсона с поправкой Йетса), ишемическая болезнь сердца у 18,37% (n=18) и у 16,49% (n=16) пациентов первой и второй групп соответственно (р=0,87622, критерий χ2 Пирсона с поправкой Йетса). Необходимо отметить, что большинство зарегистрированных в ходе исследования НЯ обусловлены проявлениями сопутствующих заболеваний и, по мнению исследователей, не имели причинно-следственной связи с изучаемыми препаратами.
На протяжении 24 недель исследуемый препарат и препарат сравнения продемонстрировали сопоставимые профили безопасности: частота развития НЯ или СНЯ в группах статистически значимо не различалась. Хотя бы одно НЯ (любой степени тяжести) или СНЯ зарегистрировано у 80,61% (n=79) пациентов первой группы и у 78,35% (n=76) больных второй (р=0,823, критерий χ2 Пирсона с поправкой Йетса), при этом связь с исследуемой терапией не исключалась у 17,35% (n=17) пациентов первой группы и у 11,34% (n=11) второй (р=0,322, критерий χ2 Пирсона с поправкой Йетса).
С клинической точки зрения наибольший интерес представляют НЯ 3–4-й степеней тяжести и СНЯ, а также оценка исследователями связи зарегистрированных НЯ и СНЯ с терапией.
Частота НЯ 3–4-й степеней тяжести была несколько выше, но статистически незначимо в группе пациентов, получавших препарат сравнения: 40,82% (n=40) и 47,42% (n=46) в группах 1 и 2 соответственно (р=0,920, критерий χ2 Пирсона с поправкой Йетса).
Подавляющее большинство НЯ 3–4-й степеней тяжести представлены повышением артериального давления и повышением уровня калия в крови. Поскольку данные изменения характерны для ХБП, в абсолютном большинстве случаев данные НЯ расценивались как не связанные с исследуемой терапией.
Связь НЯ 3–4-й степеней тяжести с исследуемой терапией отмечена у 5,1% (n=5) пациентов первой группы (во всех случаях НЯ представлены повышением АД 3-й степени тяжести) и у 6,2% (n=6) пациентов второй (у 3,1% (n=3) пациентов зафиксировано повышение АД 3-й степени тяжести, у 3,1% (n=3) – повышение уровня калия в крови 3-й степени тяжести).
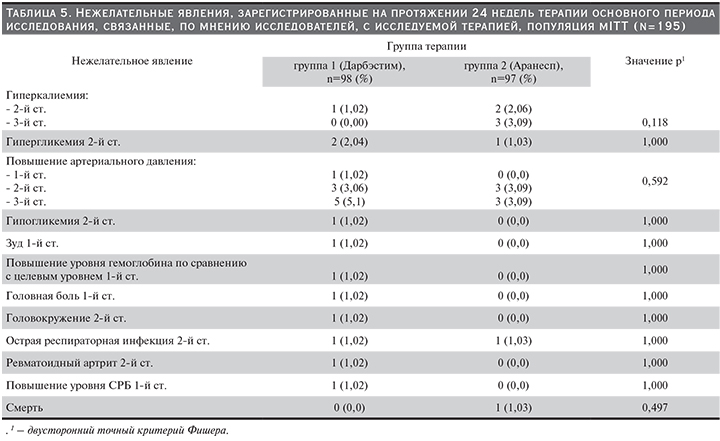
В табл. 5 представлены НЯ всех степеней тяжести, зарегистрированные в течение 24 недель основного периода исследования и связанные, по мнению исследователей, с применением препаратов дарбэпоэтина альфа.
В рамках исследования у одного пациента первой группы зарегистрирован случай развития ревматоидного артрита, сопровождавшийся повышением уровня С-реактивного белка, который, по мнению исследователей, имел возможную связь с применяемым препаратом дарбэпоэтина. Врачи-исследователи обращали внимание на то, что определенной связи, в т.ч. патогенетической, между развитием данного НЯ и проводимой терапией не прослеживается. Однако, поскольку манифестация ревматоидного артрита у данного пациента произошла вскоре после начала терапии дарбэпоэтином альфа, невозможно было полностью исключить причинно-следственную связь. В литературе не удалось обнаружить сведений о развитии ревматоидного артрита на фоне применения эритропоэзстимулирующих препаратов. Прочие НЯ, связанные, по мнению исследователей, с проводимой терапией, наблюдались в клинических исследованиях и на этапе пострегистрационного применения оригинального препарата дарбэпоэтина альфа, что указано в инструкции по медицинскому применению препарата Аранесп.
НЯ с критерием серьезности в рамках данного периода исследования зафиксированы у 12,24% (n=12) пациентов первой группы и у 10,31% (n=10) – второй (р=0,841, двусторонний точный критерий Фишера). Из них только два случая развития СНЯ (по одному случаю в каждой группе) связаны, по мнению исследователей, с терапией, статистически значимых различий между группами не было (р=1,000, двусторонний точный критерий Фишера). В группе пациентов, получавших Дарбэстим, у одного участника зафиксировано развитие артериальной гипертензии 3-й ст. с критерием серьезности «госпитализация или ее продление», исход данного СНЯ – «улучшение», степень связи с терапией – «возможная». Во второй группе зафиксирована смерть пациентки 75 лет. В рамках исследования больная получала Аранесп в течение 6 месяцев. Известно, что у пациентки имелись следующие сопутствующие заболевания: ИБС, стенокардия III ФК, стеноз аортального клапана, генерализованный атеросклероз, артериальная гипертензия, пароксизмальная форма фибрилляции предсердий, хроническая сердечная недостаточность II ФК. Причина смерти не установлена, в связи с тем что аутопсия не проводилась. В качестве предположительной причины смерти исследователем указана «острая сердечно-сосудистая недостаточность?», что является непредвиденной реакцией для препаратов дарбэпоэтина альфа. Смерть по неизвестной причине также расценивается как непредвиденная реакция. Степень причинно-следственной связи, по мнению исследователя, «возможная». Анализ данных позволяет предположить, с одной стороны, возможную декомпенсацию сердечно-сосудистой патологии, что может быть не связано с терапией дарбэпоэтином альфа (учитывая возраст пациентки, данные анамнеза, а также удовлетворительную переносимость терапии дарбэпоэтином альфа в течение полугода). С другой стороны, в данном случае нельзя исключить тромбоэмболические осложнения у пациентки, имевшей факторы риска (возраст – 75 лет, пароксизмальная форма фибрилляции предсердий), которые могут развиваться на фоне терапии дарбэпоэтином альфа. Возможны также и иные причины смерти.
Частота тромботических осложнений в первой группе составила 2,04% (n=2; тромбоз сосудистого доступа, острый инфаркт миокарда), во второй – 1,03% (n=1; тромбоз сосудистого доступа); статистически значимых различий между группами не выявлено (р=1,000). Несмотря на то что данные реакции считаются ожидаемыми при проведении терапии препаратами дарбэпоэтина альфа, по мнению исследователей, связь с лечением во всех случаях отсутствовала и развитие их было обусловлено основным заболеванием или сопутствовавшей патологией.
Также у небольшого числа пациентов зафиксированы единичные отклонения биохимических показателей, а также отклонения в общем анализе крови, включившие лимфопению, нейтропению, лейкопению. В отношении этих НЯ исследователи отмечали отсутствие причинно-следственной связи с исследуемыми препаратами. Прочие НЯ регистрировались в виде единичных случаев. Статистически значимых различий между группами по частоте зарегистрированных НЯ, не связанных с исследуемой терапией, выявлено не было.
Оценка иммуногенности
В анализ иммуногенности включены все пациенты, получившие хотя бы одно введение исследуемого препарата или препарата сравнения, у которых имелись результаты исследования на скрининге и хотя бы на одном из последующих визитов (n=192). Исследование не выявило формирования САТ ни у одного из пациентов, в связи с чем анализ на наличие НАТ не выполнялся.
Обсуждение результатов
Для создания белка, который будет использован в качестве действующего вещества в биотехнологическом препарате, используется линия живых клеток. Как правило, это обеспечивается методами генной инженерии: в клетку-продуцент внедряется чужеродная генетическая последовательность, ответственная за кодирование строго определенного белка. После того как процесс трансфекции успешно завершен, инициируются процессы по наращиванию такой модифицированной клеточной культуры и выделению из нее необходимого белка с последующей многоступенчатой очисткой и созданием готовой лекарственной формы. Таким образом, производство биотехнологического препарата – крайне сложный высокотехнологичный процесс, требующий соблюдения жестких стандартов и огромного количества требований регуляторных органов.
Биоаналоговый лекарственный препарат (биоаналог, биосимиляр, биоподобный препарат) является биологическим препаратом, схожим по параметрам безопасности, качества и эффективности с оригинальным биологическим лекарственным средством в эквивалентной лекарственной форме. В настоящее время в связи с растущим в мире интересом к применению биоаналогов объем исследований, проведение которых необходимо для внедрения таких препаратов в практическую медицину, регламентируется органами здравоохранения большинства развитых и развивающихся стран. С 1 января 2016 г. в силу вступили единые требования к лекарственным препаратам, обращающимся на рынке стран Евразийского экономического союза, гармонизированные с руководствами ВОЗ, ЕМА и FDA. Таким образом, в настоящее время требования к биоаналогам, регистрируемым в РФ, полностью соответствуют международным.
Подходы к исследованию биоаналогов, описанные в рекомендациях EMA, включают детальное сравнительное исследование физико-химических и биологических свойств, сравнительные доклинические исследования на релевантных видах животных, сравнительные исследования фармакокинетики и фармакодинамики, сравнительное исследование эффективности и безопасности [9].
Главное отличие сравнительного исследования эффективности биоаналога и референтного препарата от клинического исследования III фазы для оригинального лекарственного средства состоит в том, что его целью служит доказательство не эффективности или каких-либо иных преимуществ биоподобного препарата по отношению к плацебо или стандартной терапии, а отсутствия различий в эффективности при сравнении с референтным препаратом, поскольку ранее превосходство оригинального лекарственного средства над стандартной терапией уже было доказано.
Целью представленного исследования III фазы было доказать эквивалентную эффективность и безопасность биоаналога дарбэпоэтина альфа Дарбэстим и референтного препарата Аранесп при лечении анемии у больных ХПН. Согласно руководству EMA, одним из возможных вариантов проведения клинических исследований эффективности и безопасности (III фаза) воспроизведенных препаратов, содержащих рекомбинантные эритропоэтины, является использование параллельного дизайна с включением двух групп пациентов с анемией, вызванной ХПН, включая группы исследуемого препарата и сравнения с периодом лечения продолжительностью не менее 24 недель и общей продолжительностью исследования 1 год для оценки иммуногенности исследуемой терапии; при этом оценку иммуногенности предпочтительно проводить при подкожном введении препаратов, поскольку иммуногенность препаратов рекомбинантного эритропоэтина выше именно при подкожном пути введения [9].
В соответствии с проведенными расчетами объем выборки в данном исследовании достаточен для получения достоверных результатов.
Применявшийся в рамках данного исследования метод блочной рандомизации пациентов в группы и двойной слепой метод маскирования препаратов на протяжении всего периода терапии позволил избежать предвзятости и субъективной оценки результатов лечения. Кроме того, проведенная стратификация участников в зависимости от наличия факторов риска сосудистых тромботических осложнений (возраст ≥60 лет, наличие сосудистых протезов, наличие сахарного диабета) способствовала тому, что группы были равнозначными по количеству пациентов с вышеперечисленными признаками.
Показатель «изменение уровня гемоглобина в течение периода оценки по сравнению с исходным уровнем гемоглобина», выбранный в качестве первичной конечной точки для оценки эффективности, – один из наиболее чувствительных при оценке эквивалентности свойств биоаналога свойствам референтного препарата [9].
Таким образом, дизайн представленного исследования, объем выборки, общая продолжительность периода применения препаратов, исследуемая популяция, первичная конечная точка для оценки эффективности соответствуют международным стандартам и обусловливают высокую степень доказательности данного исследования.
Согласно полученным результатам, ДИ для разницы средних арифметических значений изменения уровня гемоглобина между двумя группами составил [-3,04; 2,93] г/л. Попадание ДИ внутрь предустановленных границ эквивалентности [-5,0; 5,0] г/л позволяет сделать вывод об эквивалентности препаратов Дарбэстим и Аранесп. Отсутствие статистически значимых различий между двумя группами терапии по таким показателям, как средний уровень гемоглобина, средняя доза дарбэпоэтина альфа, доля пациентов, которым потребовалось проведение гемотрансфузии, также свидетельствует об эквивалентной эффективности исследуемого препарата и препарата сравнения.
На протяжении исследования как препарат Дарбэстим, так и препарат Аранесп показали благоприятный профиль безопасности. В целом проведенная противоанемическая терапия переносилась пациентами удовлетворительно. Между группами не было выявлено статистически значимых различий как в спектре, так и в частоте зарегистрированных НЯ и СНЯ, в т.ч. связанных, по мнению исследователей, с терапией. С учетом исследуемой популяции и данных по безопасности референтного препарата дарбэпоэтина альфа спектр большинства зарегистрированных НЯ, связанных с терапией, ожидаем. Также необходимо отметить, что ни в одной из групп не было зарегистрировано случаев формирования САТ к дарбэпоэтину альфа, что свидетельствует о низкой иммуногенности как исследуемого препарата, так и препарата сравнения.
Таким образом, анализ методологии проведения исследования не выявил рисков ошибок, предвзятости, субъективности. С позиций доказательной медицины результаты данного двойного слепого рандомизированного исследования обладают степенью доказательности 1В [18].
Заключение
Дарбэпоэтин альфа – один из препаратов выбора для коррекции анемии у больных ХБП [19], однако его широкое применение в клинической практике в РФ ограничено высокой стоимостью оригинального препарата.
ЗАО «БИОКАД» разработан препарат Дарбэстим, являющийся биоаналогом препарата Аранесп. При его разработке учтены все требования отечественных и зарубежных регуляторов. Результаты проведенных доклинических исследований позволили сделать вывод об эквивалентности физико-химических и биологических свойств нового препарата Дарбэстим и референтного препарата Аранесп.
Результаты двух исследований I фазы с участием здоровых добровольцев подтвердили эквивалентность фармакокинетических и фармакодинамических параметров, а также показателей безопасности и иммуногенности как при однократном подкожном и внутривенном введении, так и при многократном внутривенном применении.
В рамках исследования III фазы в популяции пациентов с ХПН, находившихся на гемодиализе, проведено сравнение эффективности и безопасности препаратов Дарбэстим и Аранесп. В исследование включались пациенты, регулярно получавшие эритропоэзстимулирующие препараты, поддерживающие целевой уровень гемоглобина в течение 3 месяцев до даты рандомизации в исследование.
В течение 24 недель основного периода исследования абсолютному большинству пациентов как первой, так и второй групп удавалось поддерживать уровень гемоглобина в пределах целевых значений при применении аналогичных доз дарбэпоэтина альфа в составе как препарата Дарбэстим, так и препарата Аранесп. Профиль безопасности препаратов также не отличался.
Таким образом, результаты анализа данных, полученных в рамках основного 24-недельного этапа исследования, позволяют говорить о доказанной эквивалентной эффективности и сравнимой безопасности препаратов Дарбэстим (ЗАО БИОКАД, Россия) и Аранесп (Амджен Европа Б.В., Нидерланды).
