
В последние годы изучение нарушений правильного строения органов мочевой системы (ОМС) приобретает особое значение в связи с усиливающимися экологическими и экономическими трудностями, влияющими на развитие плода [1]. Это не значит, что до ХХ в. не существовало болезней, зависящих от нарушения формирования ребенка в антенатальном периоде его развития. Но на болезни, связанные с нарушением эмбриогенеза, не обращали должного внимания из-за неготовности медицины помочь в случае рождения ребенка с патологией генетического или тератологического генеза. Во второй половине ХХ в. внимание к почечному дизэмбриогенезу заметно возросло. В большой мере это связано с появлением методов заместительной терапии в виде диализа и почечной трансплантации. В 1971 г. R. Risdon [2] сообщил, что из 150 удаленных почек в 76 случаях имела место дисплазия. В 1970-е гг. указывалось, что почечная дисплазия может выявляться не только у детей, но и у подростков, а также у взрослых как основа, на которую наслаивается иммунная или микробно-воспалительная нефропатия [3]. О почечных дисплазиях говорили в монографиях P. Royer et al. (1973) [4], М.С. Игнатова, Ю.Е. Вельтищев (1978) [5], а Е.А. Харина и соавт. [6] дали клинико-морфологическое описание гипопластической дисплазии почек (1982), которое в современной литературе чаще фигурирует как hypoplasia/dysplasia, при которой нередко выявляется гломерулонефрит.
В настоящее время все большее значение приобретает изучение генетических влияний на формирование органов мочевой системы [7]. Можно полностью согласиться с мнением Н.П. Бочкова и его соавт. [8], высказанное еще в 1964 г., что все заболевания, кроме травм, обморожений, ожогов, укусов ядовитых животных, имеют генетическую природу. Но даже в тех случаях, когда происходит экстремальное повреждение, репарация опять-таки связана с особенностью генетической природы человека.
Говоря о дизэмбриогенезе ОМС, очевидно, следует выделять:
• анатомические изменения структуры почек и органов мочевыведения;
• паренхиматозные нарушения строения почек;
• аномалии клеточные, субклеточные, мембранозные, обычно связанные с нарушениями белковых структур и определенные состоянием биологически активных веществ, управля-
ющих их функцией;
• особую группу заболеваний, которую представляют наследственные системные болезни, патология почек при хромосомных аномалиях и эмбриональные опухоли.
Указанное подразделение на группы патологии, связанные с дизэмбриогенезом, не означает, что не наблюдается сочетанных анатомических, паренхиматозных, клеточных/субклеточных проявлений нарушения эмбриогенеза. Наоборот, разделение этих состояний условно, т. к. в клинической практике чаще встречается одновременное присутствие всех вышеуказанных изменений при дизэмбриогенезе.
Антенатальное развитие ребенка разделяется на эмбриональный и фетальный периоды. Основной органогенез мочевой системы формируется в эмбриональный период, о чем следует помнить при наблюдении за развитием беременности. Обязательно внимание и к фетальному периоду развития ребенка, т. к. инфицирование матери может сказаться на состоянии уже почти сформированных ОМС. Количество нефронов – основных действующих элементов почки останется прежним в нормальных условиях на всем протяжении жизни, если нет заболеваний, вызывающих их склерозирование и потерю функции. В последние годы выяснено, что в антенатальном периоде может закладываться меньшее число нефронов, чем это наблюдается обычно. И это малое количество нефронов нередко определяет повышенный уровень артериальное давления у взрослых [9]. Существует заболевание, при котором число нефронов ограниченно – это олигонефрония, когда гломерулы расположены далеко друг от друга в связи с их уменьшенным количеством [4].
На развитие почки в детском возрасте, у подростков и взрослых влияет фетальный объем почки. Этот вывод сделан на основании двукратного допплеровского исследования 1215 беременных женщин, в тех случаях когда в фетальном периоде отмечалось уменьшение объема почки. Следствием этого оказывается уменьшение размеров почек у родившегося ребенка, что чревато развитием нефропатий в более старшем возрасте [10]. Большое значение в развитии почки в постнатальной жизни имеет состояние подоцитов в антенатальном периоде формирования ребенка. Эти данные представлены
S.J. Shanrland et al. (2007) [11] в экспериментальной работе с культурой подоцитов, которая позволяет делать выводы о функции этих важных клеток в почке, как нейроны в головном мозгу и кардиомиоциты в сердце. Важность изучения состояния подоцитов и их щелевой мембраны в большой степени связана с тем, что мутация генов, определяющих функцию белков, входящих в состав подоцитов и шелевой диафрагмы, приводит к развитию нефротического синдрома (НС) [12, 13].
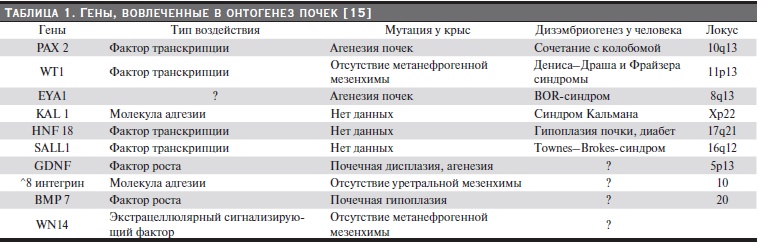
Таблица 1. Гены, вовлеченные в онтогенез почек [15].
В общей сложности в формировании ОМС участвует более 200 генов [14]. В ESPN Handbook A. Oner and R. Salamon [15] выделяют часть генов, известных с 2000 г. как причина дизэмбриогенеза у человека и причина мутации у крыс (табл. 1). В последующие годы сведения о причинах дизэмбриогенеза продолжают пополняться.
В развитии ОМС принимают участие факторы транскрипции и ростовые факторы. Большое значение в процессе формирования мочевой системы имеет апоптоз, т. к. до окончательной почки (метанефрос) у человека развивается первичная (пронефрос) и вторичная (мезонефрос) почки, клетки которых
своевременно в нормальных условиях должны элиминироватся [16]. Апоптоз весьма важен в процессе формирования окончательной почки, но некоторые факторы роста, в частности эпителиальный фактор роста и фактор роста фибробластов, защищают клетки метанефрогенной ткани от апоптоза на стадии формирования метанефроса.
Нефрогенез – сложный процесс. Образование окончательной почки происходит из двух источников: мочеточникового тяжа – производного мезонефрогенного протока, и мезенхимы метанефрогенной бластемы. Молекулярная регуляция нефрогенеза весьма сложна [17]. В ней принимает участие глиальный нейротрофический фактор (GNF). Нарушение его функции ведет к развитию диспластичной почки или ее агенезии. Большое значение в развитии окончательной почки имеет семейство генов PAX. Два из них (PAX2 и PAX8) экспрессируются в развивающейся почке с некоторым перекрытием по
времени. При мутации гена PAX2 у человека может развиться заболевание, при котором имеют место почечная дисплазия, пузырно-мочеточниковый рефлюкс (ПМР) и колобома.
Фактор транскрипции WT1 принимает активное участие в формировании половой и мочевой систем. Мутации генов WT1 приводят к развитию псевдогермофродитизма и нефротического синдрома. Сообщается также, что мутация WT1 может приводить к дизгенезии почек и развитию почечной
недостаточности у взрослых[18].
Белки семейства WNT стимулируют трансформирование метанефрогенной ткани в клетки эпителия. Гены EMX2 и BP2 в эмбриогенезе взаимодействуют с геном PAX2. Клетки, экспрессирующие ген BP2, становятся в окончательной почке интерстициальными клетками, а клетки, экспрессирующие ген
PAX2, – эпителиальными клетками. У мышей с изменениями в гене EMX2 отмечается нарушение ветвления мочеточникового выроста.
Важную роль в развитии почки имеет взаимоотношение между молекулами адгезии, в частности Е-катгерина и NCAM (молекулы адгезии нервных клеток), с гликопротеинами внеклеточного матрикса. Их взаимодействие регулируется интегринами – гетерогенными белками, образованными двумя цепями – α и β. Для развития почки существенная роль принадлежит α3β¹- и α8β¹-интегринам. В эксперименте
показано, что делеция гена α3β¹-интегрина сказывается на формировании подоцитов, базальных мембран (БМ) и ветвлении мочеточникового выроста.
Существенную роль в формировании почки оказывает интерфероноподобный фактор роста: при его блокировании невозможно в эксперименте получить развитие окончательной почки. Эпидермальный фактор роста, факторы роста фибробластов и их рецепторы участвуют в образовании канальцев. Важную роль играют ферменты: металлопротеиназы (MMP7, MMP19, TIMP1), которые влияют на деградацию матрикса, что наблюдается при почечных дисплазиях [19]. При этом отмечается уменьшение в диспластичных почках генов рецептора 2-го типа ангиотензинa II (AGTR2), глипикана 3 (GPC3) и других генов, ответственных за правильное формирование ОМС.
В последние годы не только нефрологи, но и урологи обратили внимание на развитие дизэмбриогенеза ОМС, связанного с генетическими мутациями [20]. В связи с этим хотелось бы представить номенклатуру заболеваний ОМС, которая была нами дана совместно с Ю.Е. Вельтищевым [21] в книге “Нефрология”(2000), под редакцией И.Е. Тареевой. Объединены нефро- и уропатии по той причине, что последние при их тяжелом прогрессирующем развитии обязательно вызывают
поражение почечной паренхимы, т. е. приводят к клинико-анатомическим изменениям почек с возможным последующим исходом в хроническую почечную недостаточность (ХПН).
Номенклатура наследственных и врожденных нефро- и уропатий (по М.С. Игнатовой, Ю.Е. Вельтищеву, 1989 [22], с дополнениями):
1. Нефро- и уропатии при анатомических аномалиях почек:
- количественные (агенезия, аплазия, добавочная почка, удвоение);
- позиционные (дистопия, нефроптоз, ротация);
- нарушение формы (подковообразная, L- и S-образные почки);
- изменения мочеточников, мочевого пузыря и уретры (количества, калибра, формы);
- изменения почечных сосудов (артериальных, венозных, лимфатических);
- нарушение иннервации ОМС, нередко с синдромом нейрогенного мочевого пузыря.
2. Нефропатии при паренхиматозных аномалиях почек:
- кистозные:
• поликистозная болезнь почек (варианты аутосомно-доминантные (ПКБI, ПКБII) и АРПКБ);
• комплекс медуллярных кистозных болезней, включающих нефронофтиз и кистозы в сочетании с другими аномалиями;
• кистозные дисплазии;
• гломерулярные кистозы;
• др.;
- бескистозные:
• олигонефрония, олигомеганефрония;
• сегментарная гипоплазия (болезнь Аск–Упмарка);
• при гипопластической дисплазии почек;
- рефлюкс-нефропатия (сочетание врожденного ПМР нередко
одновременно с дисплазиями различных участков ОМС).
3. Нефропатии при клеточных, субклеточных, мембранных аномалиях почек:
- наследственный нефрит (Синдром Альпорта);
- болезнь тонких базальных мембран;
- тубулопатии первичные:
• с преимущественным поражением проксимальных канальцев (болезнь и синдром де Тони–Дебре–Фанкони, глицинурия, цистинурия, фосфат-диабет и др.);
• с преимущественным поражением дистального канальца (почечный тубулярный ацидоз I типа, нефрогенный несахарный диабет, псевдогипоальдостеронизм);
• с повреждением всего канальцевого аппарата;
• с нарушением реабсорбции натрия в эпителиальном натриевом канале кортикальной части собирательных трубок (с ранним развитием артериальной гипертензии (синдром Лиддла, гиперальдостеронизм и др.) и артериальной гипотензии (синдромы Барттера, синдром Гительмана);
- тубулопатии вторичные при наследственной патологии обмена веществ:
• дизметаболические нефропатии с кристаллуриями;
• оксалатная нефропатия (наследственная первичная гипероксалурия I, II, III типов);
• дизметаболическая нефропатия с оксалатно-кальциевой кристаллурией;
• уратная (подагрическая) нефропатия.
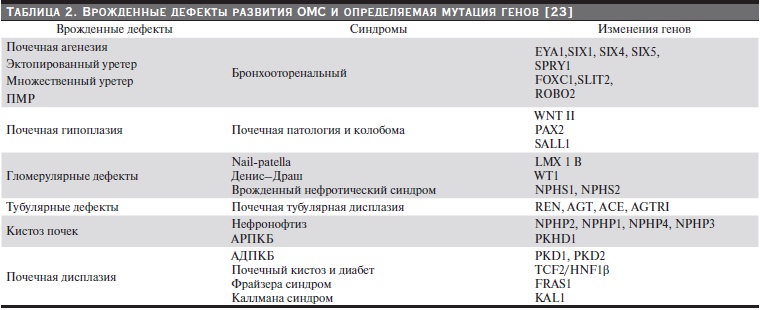
Среди анатомических аномалий ОМС преобладают случаи врожденных пороков развития почек и органов мочеотделения, которые носят название CAKUT. В это понятие включен большой спектр аномалий в виде гипоплазии, дисплазии почек, аномалий уретры в виде мегауретера и удвоения уретры, а также другие проявления нарушения антенатального развития ОМС. Встречается в 50 % случаев у новорожденных с патологическими изменениями в брюшной полости [23] и у 1 из 500 детей, которым проводится УЗИ в фетальном периоде развития ребенка [24] . Естественно, эта патология чревата поражением почечной паренхимы и развитием нефропатии и ХПН. Причем CAKUT может быть изолированным патологическим синдромом, но в ряде случаев он сочетается с аномалиями других органов. При этом известны или подозреваются различные гены в их развитии (табл. 2). Известно, что начало формирования ОМС у человека происходит с 22-го дня гестации. На основании экспериментальных исследований на мышах кандидатом в гены, определяющие процесс формирования CAKUT, включен ген, контролирующий белок ESRRG-рецептор стероидных гормонов [25].
В последние годы большое внимание уделяется дизэмбриогенезу паренхимы почек, прежде всего поликистозным заболеваниям и нефронофтизу. Это в определенной мере связано с достижениями молекулярной биологии и генетики в плане изучения роли цилий в развитии этих заболеваний
[26]. Существует два вида цилий (ворсинок, ресничек). Один вид – подвижные цилии, заболевания, связанные с их аномалиями, интересует в основном пульмонологов и оториноларингологов, а характерным представителем этой патологии является синдром Картагинера. В последние годы (конец ХХ и начало ХХI в.) обращено внимание на другой вид цилий, называемых первичными цилиями (неподвижные ворсинки), или моноцилиями, которые представлены на поверхности эпителиальных клеток практически всех органов млекопитающих. Определено, что моноцилии функционируют как сенсорные органеллы, обеспечивающие выработку рецепторных молекул и инициирующие ток жидкости, в частности, вокруг эмбрионального узла. Реснички весьма динамичны в плане обновления собственной структуры, которая происходит с участием белков-кинезинов, способствующих перенесению структурных белков цилии из цитоплазмы к микротрубочкам, за счет чего происходит структурная сборка белков этой органеллы. Аксонема – это основная структурная единица цилий, которая состоит из 9 дуплетов (пар) микротрубочек, У подвижных цилий – 9 + 2 микротрубочки, 9 из которых расположены вокруг центральной пары. Неподвижные цилии (9 + 0) не имеют центральной пары (рис. 1). Дисфункция белков цилий проявляется в фенотипе болезней органной специфичности, таких как поликистозная болезнь, и при полиорганных синдромах, таких как болезнь Bardet–Biedle
[27]. Дефекты цилий отмечены в развитии поликистозной болезни почек, печени, поджелудочковой железы, дефектах неврогенной трубки, полидактилии, situs inversus, дегенерации сетчатки. В настоящее время доказано, что нефронофтиз является цилиопатией, при которой в связи с отсутствием нефроцистина-3 наблюдаются изменения белков цилий с развитием тяжелой полиорганной патологии вплоть до эмбриональной летальности [28]. Еще не известно, каким
образом цилии вызывают дизэмбриогенез, но уже ясно, что леворасположение органов, кистозы почек, печени и поджелудочковой железы, а также ожирение связаны с дефектами этих органелл [29]. Пристальное внимание к кистозам связано с тем, что в состав аксонем неподвижных цилий входят практически все белки, мутация генов которых осуществляется при развитии поликистозных болезней (аутосомнодоминантных и аутосомнорецессивных) и нефронофтизе.
Таблица 2. Врожденные дефекты развития ОМС и определяемая мутация генов [23].
Кроме изучения анатомического и паренхиматозного дизэмбриогенеза бальшое внимание уделяется аномалиям развития на клеточном/субклеточном и мембранном уровнях. К ним относятся разнообразные тубулопатии, развитие которых, как правило, связано с аномалиями белковых структур канальцев почек, обусловленных генными мутациями [30]. К этой группе патологии относятся наследственный нефрит и болезнь тонких базальных мембран (БТБМ). Исследования мутантных генов в
1970–1980 гг. как основы развития нефрологической патологии только начинались, и мы [31] причину наследственного нефрита и гипопластической дисплазии почек изучали на основании определения экскреции оксилизингликозидов с мочой. В работе R.Williamson et al. (1997)[32], которые исследовали состояние дистрогликанов в эксперименте на эмбрионах мышей, указано на резкие изменения содержания дистрогликанов в организме экспериментальных животных при наличии почечной диспла-
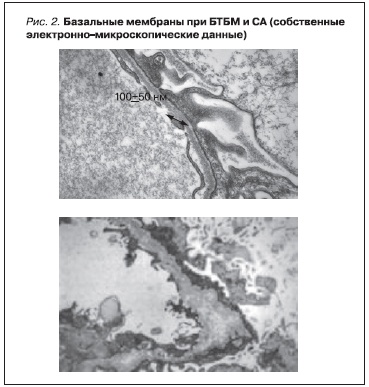
зии. В последние годы большое внимание уделяется состоянию IV фракции коллагена при различных вариантах дизэмбриогенеза; преимущественно исследуются больные с синдромом Альпорта (СА) и БТБМ. При этих заболеваниях молекулярно-генетически они и могут связываться с мутацией одних и
тех же генов [33], но морфологические проявления различны: своеобразное утолщение и специфические изменения БМ при СА и ее диффузное истончении при БТБМ (рис. 2).
Рисунок 1. Морфология цилии [28]
Рисунок 2. Базальные мембраны при БТБМ и СА (собственные электронно-микроскопические данные).
Дизэмбриогенез ОМС чаще всего начинает проявлять себя в раннем детском возрасте. Поэтому, если под наблюдение попадает ребенок новорожденный или первых месяцев жизни, думая о сути его заболевания, нужно помнить о возможности дизэмбриогенеза наследственного или связанного с тератогенными факторами.
У новорожденных и младенцев первых месяцев жизни прежде всего проявляются врожденные и тяжелые генетически обусловленные синдромы, чаще это пороки развития различных структур ОМС: от грубых анатомических аномалий, не совместимых с жизнью ребенка, до изменений на клеточном
или субклеточном уровне, хотя последние в первые месяцы жизни ребенка могут и не проявляться. Исключение составляет финский тип врожденного НС. Проявления последнего могут присутствовать у ребенка еще до его рождения. В первые дни–недели жизни ребенка могут ярко манифестировать
болезни в виде сложных генетических синдромов, где почки являются одним из органов системного дизэмбриогенеза. Врожденные и наследственные заболевания у новорожденного ребенка, существующие изолированно или в сочетании с наслоением инфекции мочевой системы, обычно приводят к критическому состоянию, когда одна или несколько жизненно важных функций организма утрачены полностью или требуют существенной поддержки [34].
Изучение структуры и частоты заболеваний ОМС у новорожденных в Московской специализированной клинике больницы им. Н.Ф. Филатова показало, что острая почечная недостаточность (ОПН) составляет примерно 16 % от всей выявляемой у этой группы детей патологии [35]. Развивается
ОПН, как правило, при наличии анатомической или паренхиматозной аномалии строения почки или органов мочевыведения. В последние годы все чаще приходится сталкиваться с развитием рефлюкс-нефропатии (РН) у детей первых месяцев жизни.
Факторы риска развития РН у детей раннего возраста:
• высокие и двусторонние ПМР;
• неправильное формирование мышечной стенки лоханки;
• повышение внутрилоханочного давления;
• развитие интраренального рефлюкса;
• дисфункция сосочкового аппарата.
Все это в совокупности с инфекцией мочевой системы (ИМС) приводит к развитию рефлюкс-нефропатии [36]. В ХХI в. показано, что для развития РН необязательно должны быть повторные случаи ИМС у ребенка, достаточно наличие дисплазии не только в мочеточниковом соустье, но в других отделах мочевого тракта. Изучается роль генов ROBO2 в развитии пузырно-мочеточникового рефлюкса и CAKUT [37]. Специальное внимание должно быть уделено патологии, которая редко, но выявляется у мальчиков – клапан задней уретры. Эта аномалия строения нижных мочевых путей должна обязательно проверяться, т. к. грозит тяжелыми паренхиматозными изменениями почек.
Проявления дизэмбриогенеза почек возможны как изолированное страдание, так и как синдром при нарушении развития различных органов и систем. В некоторых случаях нефрологическая патология оказывается ведущей и на нее обращается большее внимание, чем на состояние других органов. Часто забывается, что основой неблагополучия является нарушение формирования не одного органа, а состояние дизэмбриогенеза организма в целом. Зная, что нарушение эмбриогенеза может
проявляться не только в раннем возрасте, разрабатываются программы наблюдения за развитием детей даже с нерезко выраженными аномалиями ОМС, которые предусматривают изучение причин, ведущих к уменьшению количества нефронов в антенатальный период развития. Обращается внимание на течение беременности, нарушения питания, дефицит витамина А, обструктивные и рефлюксирующие нефропатии, нефротоксические лекарства [38]. Обязательно при обследовании ребенка обращать внимание на внешние проявления соединительнотканного дизэмбриогенеза, часто называемого малыми аномалиями развитии (МАР), массоростовые показатели, наличие аномалий строения не только органов мочевой системы, но и сердца, костной системы и др., где аномалии
присутствуют, но нерезко выражены.
Чаще при почечном дизэмбриогенезе снижение функции имеет тубулярный характер, и лишь при нарастании паренхиматозных изменений снижается СКФ. Совершенно справедливо поставлен вопрос: что мы знаем о развитии нефропатии от периода новорожденности до взрослого состояния [39]?
Это тем более важно, что в настоящее время по инициативе Американского нефрологического общества во всем мире интенсивно изучается развитие хронической болезни почек (ХБП) [40].
Заключение
Таким образом, несмотря на то что нет статистики, которая указывала бы на увеличение случаев почечного дизэмбриогенеза в настоящее время, однако неблагоприятные изменения экологического и экономического состояния во всем мире предполагают увеличение случаев аномалий антенатального
развития ребенка. В большой мере это может касаться почек и органов мочевыведения. Дизэмбриогенез почек как интегрального органа, обеспечивающего постоянство физико-химического состояния крови, на анатомическом, паренхиматозном и клеточно/субклеточном уровнях даже без наслоения иммунных или микробно-воспалительных процессов может приводить к тяжелым состояниям у ребенка. Остается вопрос: повинно ли скрытое течение почечного дизэмбриогенеза в формировании ХБП у взрослых? На этот вопрос, по-видимому, ответят последующие исследования.



{kind=link}
{kind=link}
{kind=link}
{kind=link}