
Множественная миелома (ММ) – злокачественное лимфопролиферативное заболевание, характеризующееся инфильтрацией костного мозга клональными плазматическими клетками, секрецией моноклонального иммуноглобулина, выявляемого в сыворотке крови и/или моче (М-протеин) и поражением органов мишеней (почки, кости).
Эпидемиология
ММ располагается на 23-м месте в ряду наиболее часто встречающихся злокачественных новообразований, в общемировой популяции составляя 0,8% всех впервые диагностируемых опухолей и 1,0% случаев смерти, связанных с раком [1]. Заболеваемость MM отличается в зависимости от страны, расы и пола и составляет примерно 10–15% случаев гемобластозов, являясь второй по распространенности опухолью гематологической природы [2]. Заболеваемость в США в среднем составляет 4–5 новых случаев, а среди популяции афроамериканцев может достигать 9–10 случаев на 100 тыс. населения в год [3]. Напротив, в странах Восточной Азии, например в Японии, заболеваемость существенно ниже и не превышает 1,2 на 100 тыс. населения в год [4]. В 2016 г. в России было диагностировано 3848 новых пациентов ММ, общий показатель заболеваемости составил 2,6 случая на 100 тыс. населения. Максимальная заболеваемость (12,2 случая на 100 тыс. населения) зарегистрирована в когорте людей в возрасте от 70 до 74 лет [5]. Соотношение заболевших мужчин и женщин в Северной Америке и странах Западной Европы составляет 1,0/0,7–0,8, в Российской Федерации – 1,0/1,4 [2, 6]. Доступность новых методов лечения ММ привела к улучшению общей выживаемости (ОВ). Согласно данным одного популяционного регистра, 5-летняя ОВ пациентов ММ в России в период с 1994 по 2011 г. фактически увеличилась в 2 раза – с 18 до 36%, медиана ОВ – с 28 до 38 месяцев [7].
Вопросы терминологии
ММ предшествует бессимптомная незлокачественная стадия, получившая название моноклональной гаммапатии неясного значения (МГНЗ), которая представляет собой предопухолевое заболевание с ограниченным риском трансформации в симптоматическую миелому [8].
В части случаев прослеживается промежуточная стадия – тлеющая множественная миелома (ТММ), биологически располагающаяся между МГНЗ и ММ МГНЗ ассоциируется с риском прогрессирования в ММ или близкие к ней опухоли за первый год наблюдения приблизительно на уровне 1–2%, ТММ – около 10–20% [8, 9]. МГНЗ и ТММ протекают бессимптомно и, как правило, диагностируются случайно, когда при лабораторном обследовании по поводу какого-либо другого состояния обнаруживается секреция моноклонального парапротеина. В рутинной практике пациенты с МГНЗ и ТММ подлежат только динамическому наблюдению, а терапию начинают после трансформации в симптоматическую ММ [9]. Объем обследования пациентов с МГНЗ и ТММ предусматривает физикальный осмотр, общий и биохимический анализы крови, обязательно включающие определение уровня общего белка, белковых фракций, кальция и креатинина, иммунохимическое исследование белков сыворотки крови и мочи. При МГНЗ первое динамическое обследование делают через 3 месяца и, убедившись в отсутствии прогрессирования, далее каждые 12, при ТММ – каждые 3–6 месяцев. Рентгенологическое обследование костной системы – ежегодно.
До недавнего времени для верификации активной (симптоматической) ММ, нуждающейся в незамедлительном начале лечения, требовалось подтверждение специфического поражения конечных органов мишеней. Терапию начинали при обнаружении любого из четырех признаков симптомокомплекса CRAB, включающего гиперкальциемию (Calcium), почечную недостаточность (Renal failure), анемию (Anemia) и остеолитическое поражение костей (Bone lesions). Подобная тактика призвана оградить пациентов с МГНЗ и ТММ от преждевременного начала токсичной химиотерапии. До недавнего времени не было ни одного клинического исследования, которое бы подтвердило преимущество начала терапии на стадии ТММ. Тем не менее для пациентов ТММ высокого риска характерно ограниченное время до трансформации и не разумно дожидаться, когда у пациента вдруг разовьются повреждение почек или костные осложнения. Таким образом, необходимо было идентифицировать биологические маркеры, которые могли бы помочь отличить истинно индолентные процессы от состояний, которые в течение короткого отрезка времени презентируют с классического симптомокомплекса CRAB.
Пересмотренные диагностические критерии ММ
В 2014 г. экспертами Международной рабочей группы по множественной миеломе (IMWG) были пересмотрены диагностические критерии ММ [10]. Новые критерии ММ позволяют использовать предложенные биомаркеры, чтобы определить показания к началу лечения в отсутствие у пациента традиционных критериев CRAB (табл. 1).

События, определяющие множественную миелому
Морфологическая верификация ММ предполагает необходимость диагностировать ≥10% клональных плазматических клеток в аспирате костного мозга либо подтверждение с помощью гистологического исследования, например биоптата плазмоцитомы. Помимо этого необходимо наличие одного или более событий, определяющих миелому. Под таковыми понимают наличие одного или более признаков CRAB или одного или более маркеров биологической злокачественности. В определение ММ включено
3 биомаркера, которые ассоциируются приблизительно с 80% риском раннего прогрессирования в симптоматическую миелому с поражением органов-мишеней. В качестве последних предлагается учитывать (1) количество клональных плазматических клеток в костном мозге ≥60%, (2) соотношение между вовлеченными и невовлеченными свободными легкими цепями иммуноглобулинов (СЛЦ) ≥100 при условии, что уровень секреции вовлеченной СЛЦ составляет не менее 100 мг/л и/или (3) верификацию более чем одного очага поражения костного мозга по данным МРТ [10].
Экстремальный плазмоцитоз костного мозга
Необычайно высокая инфильтрация костного мозга с количеством клональных плазматических клеток ≥60% в отсутствие симптомов CRAB встречается крайне редко. В исследовании клиники Mayo (1996–2010) только у 3,2% пациентов с ТММ (n=651) имела место подобная картина костного мозга. В течение 2 лет наблюдения трансформировались в симптоматическую ММ до 95% пациентов этой группы [11]. Похожие результаты показали греческие исследователи, диагностировавшие инфильтрацию костного мозга ≥60 у 8% пациентов с ТММ (n=96). Медиана времени до прогрессирования (ВДП) в этой подгруппе составила 15 месяцев против 90 (р<0,001) в случае плазмоклеточной инфильтрации <60% [12].
Соотношения между вовлеченными и невовлеченными СЛЦ
При ТММ нарушение соотношения между СЛЦ также ассоциируется с повышенным риском прогрессироания. В исследовании клиники Mayo (n=586; 1970–2010) изменение соотношения между вовлеченными и невовлеченными СЛЦ ≥100 наблюдалось у 15% пациентов с ТММ. Для этих пациентов в первые 2 года риск прогрессирования в симптоматическую ММ или AL-амилоидоз составил 79% [13]. Сходные находки представили греческие исследователи, выявившие соотношение вовлеченных и невовлеченных СЛЦ ≥100 у 7% пациентов с ТММ (n=95). Медиана ВДП для них составила 8 месяцев против 73 (р=0,001) для пациентов с индексом <100 [12]. Таким образом, значительное изменение соотношения СЛЦ в пользу секреции вовлеченной цепи служит точным предиктором высокого риска скорого прогрессирования в симптоматическую ММ. Эксперты IMWG также добавили требование, согласно которому минимальный уровень вовлеченной СЛЦ должен быть не 100 мг/л, чтобы этот критерий можно было рассматривать в качестве события, определяющего миелому [10].
Один или более литических очагов в костях по данным МРТ
МРТ считается идеальным методом диагностики мягкотканых экстрамедуллярных поражений, выявления очагов лизиса в костях позвоночника и таза. Применительно к ТММ с помощью технологии МРТ всего тела можно выявить очаговые или диффузные изменения костного мозга, которые не видны на КТ или рентгеновских снимках. По данным радиологов Гейдельбергского университета, в Германии один или более очагов поражения по данным МРТ всего тела присутствовал у 15% пациентов с ТММ (n=149). Медиана ВДП для этой группы составила 13 месяцев, а частота прогрессирования за 2 года – 70% [14]. Эти результаты полностью подтверждаются работой греческой миеломной группы. Исследователи нашли один или более литических фокусов при МРТ позвоночника у 14% пациентов с ТММ (n=65). При этом медиана ВДП была 15 месяцев и за 2 года прогрессировали в ММ 69% больных [15]. Согласно рекомендациям IMWG, диагностическое значение имеют очаги поражения размером от 5 мм и более [10]. Пациентам с единичным очагом поражения и в ситуации неоднозначной интерпретации картины рекомендуется динамическое наблюдение и повторное выполнение МРТ через 3–6 месяцев.
Современные методы визуализации поражения костей и плазмоцитом
Ранее (критерии IMWG, 2003) предполагалось, что для диагностики поражения костей как одного из симптомов CRAB будет использоваться исключительно стандартная рентгенография. Новые технологии медицинской визуализации в последнее время стали широко доступными и могут активно применяться для диагностики костных и экстрамедуллярных поражений. Все новые методы обладают большей чувствительностью по сравнению со стандартной рентгенографией костей. Диагностические возможности сканирования всего тела с помощью МРТ выше в 1,12–1,82, низкодозной КТ – в 1,04–1,33, позитронно-эмиссионной томографии (ПЭТ) с 18F-фтордезоксиглюкозой – в 1,00–1,58 и совмещенной процедуры ПЭТ/КТ – в 1,27–1,45 раза. КТ и МРТ обладают близкой чувствительностью и позволяют обнаруживать до 80% остеолитических очагов [16].
Основываясь на этих данных, пересмотренные критерии IMWG допускают для диагностики поражений при ММ использование МРТ всего тела, низкодозной КТ всего тела и ПЭТ/КТ. Обнаружение одного или более остеолитических очагов размером ≥5 мм позволяет рассматривать ситуацию как специфическое поражение костей. Изолированное накопление глюкозы при ПЭТ без доказательств реальной деструкции кости по данным КТ считается неадекватным. В такой ситуации следует обсуждать биопсию одного из очагов поражения кости, поскольку существуют сомнения в диагнозе. Согласно рекомендациям IMWG, наличие остеопороза, компрессионных переломов тел позвонков или изменений, обнаруженных с помощью денситометрии в отсутствие литических очагов не являются доказательством поражения костей при ММ [10].
Выбор метода визуализации зависит от конкретной клинической ситуации и технической доступности. Однозначно можно сказать, что диагноз ТММ или солитарной плазмоцитомы не может быть установлен только с помощью обычной рентгенографии. Минимально допустимый объем обследования для клиник с ограниченными ресурсами должен включать рентгенографию всех костей и МРТ (или КТ) позвоночного столба и таза [10].
Критерии почечной недостаточности
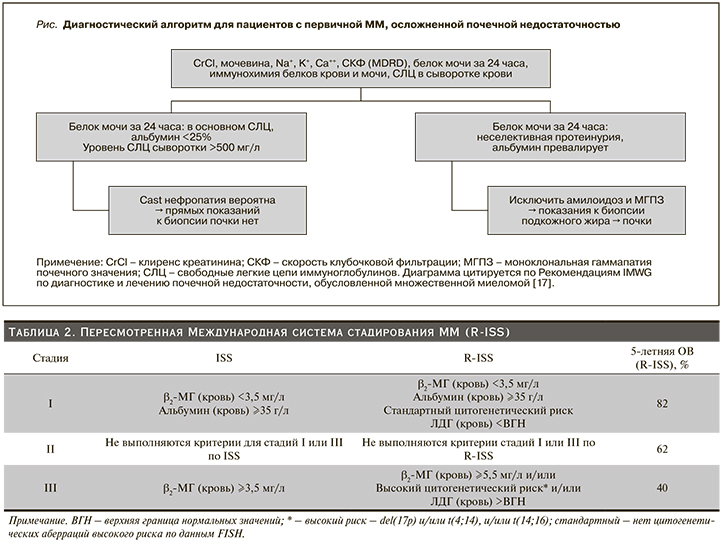
В рекомендациях IMWG предложена новая детализация термина почечной болезни (как симптома CRAB), которая теперь подразумевает в качестве такового только вероятную или доказанную цилиндровую (cast) нефропатию, обусловленную избыточной секрецией СЛЦ иммуноглобулинов [10]. Другие варианты поражения почек, обусловленные секрецией моноклонального парапротеина, такие как болезнь накопления легких цепей, мембранопролиферативный гломерулонефрит и AL-амилоидоз, рассматриваются как отдельные заболевания, не относящиеся к ММ [10]. Диагностический алгоритм анализа почечной недостаточности у пациентов впервые диагностированной множественной миеломой (ВДММ) представлен на рис. 1. Биопсия почки для уточнения этиологии почечной недостаточности рекомендуется пациентам с предполагаемой cast-нефропатией в ситуации, когда уровень вовлеченных СЛЦ в сыворотке крови ниже 500 мг/л [17]. В случае неселективной протеинурии в первую очередь следует исключать AL-амилоидоз. Рекомендуется окраска конго красным биоптата подкожно-жировой клетчатки, слизистых, трепанобиоптата костного мозга и реже непосредственно почки.
При оценке критериев CRAB следует ориентироваться на расчетную скорость клубочковой фильтрации менее 40 мл/мин (формулы CKD-EPI или MDRD), нежели просто на повышение уровня креатина сыворотки крови [10].
Солитарная плазмоцитома
Солитарная плазмоцитома – это ранняя стадия плазмоклеточной злокачественной опухоли, которая занимает промежуточное положение между ТММ и ММ. Диагноз подтверждается морфологически биопсией непосредственно плазмоцитомы кости или экстрамедуллярной опухоли в отсутствие поражения костного мозга [10]. Естественно, у пациента не должно быть никаких специфических изменений, как минимум при рентгенографии костей скелета и МРТ (или КТ) позвоночного столба и таза, за исключением собственно этой солитарной плазмоцитомы. Симптомы CRAB отсутствуют. Лечение предусматривает проведение только локальной лучевой терапия в СОД 40–50 Гр, что обеспечивает очень хорошие результаты [18]. Химиотерапия проводится при резистентности к лучевой терапии [19].
Пациентам с солитарной плазмоцитомой, которым диагностировано ограниченное поражение костного мозга (<10% клональных плазматических клеток), выставляют диагноз солитарной плазмоцитомы с минимальным вовлечением костного мозга [10]. Иммунофенотипирование в данной ситуации обязательно ввиду очевидного пограничного количества клональных плазматических клеток в костном мозге. Лечение аналогично таковому при солитарной плазмоцитоме. Однако если риск рецидива или прогрессирования в симптоматическую ММ за 3 года для пациентов с солитарной плазмоцитомой составляет приблизительно 10%, то в случае солитарной плазмоцитомы с минимальным вовлечением костного мозга уже 20–60% [19]. Проблема терапии солитарной плазмоцитомы с минимальным вовлечением костного мозга не решена. Показания к химиотерапии в этой ситуации шире.
Клиническое стадирование множественной миеломы
ММ – гетерогенное заболевание, при котором ОВ может колебаться от нескольких месяцев до 10 лет и более [20]. Существуют значительные различия по выживаемости в зависимости от физического состояния, опухолевой массы, биологии (цитогенетический риск) и ответа на терапию. Системы клинического стадирования Durie-Salmon (DSS) и Международная система стадирования (ISS) традиционно строятся на косвенной оценке опухолевый массы и не учитывают биологическое поведение [21]. DSS имеет очевидные проблемы с воспроизводимостью хотя бы потому, что необходима индивидуальная интерпретации рентгенологической картины поражения костей. ISS более воспроизводима, но включает такой фактор, как альбумин сыворотки крови, который собственно не зависим от ММ.
Биология ММ во многом определяется молекулярным вариантом болезни, наличием или отсутствием специфических цитогенетических аберраций. Стандартное кариологическое исследование при ММ обладает ограниченной чувствительностью. Оптимальным методом обследования является проведение FISH-тестов на предварительно выделенных из аспирата костного мозга плазматических клетках. Такие поломки, как t(4;14), t(14;16), t(14;20), +1q21, del(1p) и del(17p), в большинстве исследований ассоциировались с высоким риском прогрессирования и ограниченной выживаемостью [22].
В 2015 г. IMWG предложена пересмотренная Международная система стадирования (R-ISS), в которой в дополнение к ранее использовавшимся параметрам стали учитывать наличие цитогенетических аберраций высокого риска и повышение активности ЛДГ в сыворотке крови, что представляет собой оценку биологии заболевания (табл. 2). R-ISS была разработана на основании анализа прогноза 4445 пациентов с ВДММ, участвовавших в 11 международных исследований [23]. В этом исследовании показано, что 5-летняя ОВ пациентов ММ по R-ISS хорошо воспроизводима и составляет 82; 62 и 40% для I, II и III стадий соответственно.
Терапия пациентов с впервые диагностированной множественной миеломой, которым может быть выполнена трансплантация
В 2017 г. Европейским обществом медицинских онкологов (ESMO) опубликованы практические рекомендации по диагностике, лечению и мониторингу пациентов ММ [24]. С практических позиций к группе высокого цитогенетического риска рекомендовано относить пациентов при обнаружении с помощью FISH любой из трех наиболее важных аберраций, включая del(17p) и/или t(4;14) и/или t(14;16). Всех остальных пациентов следует классифицировать по категории стандартного риска.
Пациенты стандартного цитогенетического риска
Всем пациентам с ВДММ, которых рассматривают в качестве кандидатов (до 65 лет в России и большинстве стран Европы и до 75 лет в США) для аутологичной трансплантации стволовых гемопоэтических клеток (ауто-ТГСК), следует проводить от 4 до 6 циклов индукционной терапии с целью максимально возможного уменьшения опухолевой массы и купирования симптомов. В качестве индукционных режимов в России наиболее часто используют бортезомиб-содержащие программы VCD (бортезомиб, циклофосфамид, дексаметазон), PAD (бортезомиб, доксорубицин, дексаметазон) и RVD (леналидомид, бортезомиб, дексаметазон). Следующим этапом после индукции служит сбор стволовых клеток и проведение высокодозной терапии (ВДТ) с последующей ауто-ТГСК. Целью сбора стволовых клеток является забор материала для проведения по крайней мере двух трансплантаций. Под ВДТ большинство клиник рассматривают введение мелфалана в дозе 200 мг/м2 [24]. Дозу препарата снижают до 140 мг/м2 ослабленным пациентам и в случае выраженного нарушения функции почек [17].
Преимущество проведения аутоТГСК в рамках первой линии терапии по сравнению с трансплантацией после рецидива (вторая линия) продемонстрировано в большом рандомизированном исследовании IFM/DFCI 2009 (n=700), где медиана выживаемости без прогрессирования (ВБП) составила для двух разных подходов 50 и 36 месцев (р<0,001) соответственно [25]. Несмотря на то что не было получено разницы в ОВ, очевидно, что далеко не каждый пациент после терапии рецидива может реализовать эту важную опцию. Для пациентов, достигших очень хорошей частичной (ОХЧР) или полной ремиссии (ПР), обсуждается несколько стратегий, включая одну, или тандемную, ауто-ТГСК, консолидацию и поддерживающую терапию [26].
Поддерживающая терапия леналидомидом после ауто-ТГСК представляется критически важной опцией, пролонгирующей как ВБП, так и ОВ. По данным мета-анализа трех самых больших проспективных контролируемых исследований, при медиане наблюдения 79,5 месяцев за выжившими пациентами в группе леналидомида медиана ОВ не была достигнута против 86,0 месяцев(HR=0,75; 0,63–0,90; р=0,001) в группе плацебо [27]. Продолжительность поддерживающей терапии леналидомидом в Российских клинических рекомендациях ограничена двумя годами [6].
Пациенты высокого цитогенетического риска
Лечение пациентов высокого цитогенетического риска представляет собой нерешенную задачу. Текущая стратегия предусматривает включение таких пациентов в клинические исследования новых препаратов (даратумумаб, элотузумаб, карфилзомиб, иксазомиб), которые пока не одобрены для первой линии терапии. С практических позиций в качестве индукционного режима следует рассматривать комбинацию ингибитора протеасомы и иммуномодулятора (RVD) с последующей ВДТ и аутоТГСК [20, 25]. Данные по значимости двойной трансплантации пациентам высокого риска противоречивы. Так, в исследовании EMN02/H095 показано, что тандемная аутоТГСК значительно улучшает ВБП пациентов с t(4; 14) и/или del(17p) HR=0,42 (95% ДИ – 0,21–0,84; р=0,014) и ОВ по всем пациентам HR=0,52 (0,31–0,86; р=0,011) [28]. И хотя, например, в американском исследовании StaMINA не получено таких различий [29], общий тренд складывается в пользу двойной трансплантации.
Трансплантация аллогенного костного мозга может рассматриваться в отношении молодых пациентов высокого риска, но данные достаточно противоречивы и ограниченны, поскольку нет соответствующих рандомизированных исследований.
Пациентам высокого цитогенетического риска после ауто-ТГСК рекомендуется поддерживающая терапия бортезомибом в течение 2 лет [20].
Терапия пациентов, которые не рассматриваются в качестве кандидатов для трансплантации
Критерии непригодности для трансплантации являются четко не определены и не абсолютны. Обычно клиницисты принимают во внимание возраст старше 65 лет, высокий индекс коморбидности, снижение когнитивных возможностей, плохое физическое состояние, наличие старческой астении и т.п. характеристики. Выполнение ВДТ и аутоТГСК пожилым и ослабленным пациентам сопряжено с риском чрезмерной токсичности и ранней летальности [26].
К наиболее часто используемым в нашей стране режимам терапии первой линии для пациентов, не рассматриваемых в качестве кандидатов для трансплантации, относятся бортезомиб-содержащие комбинации VD (бортезомиб, дексаметазон) и VMP (бортезомиб, мелфалан, преднизолон). Комбинация на основе леналидомида (схема Rd) в качестве первой линии терапии в России зарегистрирована лишь в 2017 г., поэтому используется реже. Цель терапии пациентов с ВДММ, не пригодных для трансплантации, заключается в достижении максимально возможного глубокого ответа, поскольку в этой категории пациентов он также транслируется в лучшие показатели выживаемости. Вопрос о токсичности отдельных терапевтических схем для пожилых пациентов может быть критичным. Вопрос, какие режимы являются оптимальные на основе двух или трех противоопухолевых препаратов, остается открытым.
В качестве примера в американском исследовании UPFRONT (фаза IIIB) было выполнено сравнение трех бортезомиб-содержащих режимов (VD, VTD, VMP) в когорте довольно пожилых пациентов (медиана возраста – 73 года). В этой работе не было выявлено никаких преимуществ тройных комбинаций (VTD, VMP) по сравнению с двойной (VD). Получается, что более высокая интенсивность лечения пожилых пациентов не всегда транслируется в лучшую выживаемость [27].
Согласно данным исследования FIRST (фаза III), непрерывная терапия леналидомидом (программа Rd) обеспечивает несомненное преимущество в 4-летней ВБП по сравнению с 18 циклами Rd и 12 – MPT (32,6% против 14,3 и 13,6%; р<0,00001). Наибольший выигрыш получили пациенты, на программе Rd достигшие как минимум ОХЧР или лучшего ответа. В этой подгруппе время до следующей линии терапии составило 69,5 месяцев против 39,9 и 37,7 соответственно [30]. Возможно, что пациенты с более слабым ответом после 18 циклов Rd могут получить выигрыш в выживаемости при смене терапии на альтернативную комбинацию VMP [31].
Заключение
В 2014 г. IMWG были обновлены диагностические критерии ММ и связанных с ней заболеваний. В дополнение к симптомокомплексу CRAB в качестве критериев ММ, требующей начала терапии, были выделены 3 признака биологической злокачественности: ≥60% клональных плазматических клеток в костном мозге; соотношение вовлеченных и невовлеченных СЛЦ ≥100 и/или наличие более чем одного очага поражения костного мозга при МРТ исследовании. Низкодозная КТ всего тела, МРТ, ПЭТ и ПЭТ/КТ более чувствительны в определении ММ и должны применяться как на диагностическом этапе, так и при оценке эффективности лечения. Под поражением почек при ММ подразумевается только cast-нефропатия (цилиндровая нефропатия, обусловленная перегрузкой легкими цепями иммуноглобулинов), которая рассматривается как критерий CRAB. В 2015 г. предложена пересмотренная система стадирования R-ISS, предусматривающая учет молекулярных аберраций высокого цитогенетического риска и активности ЛДГ сыворотке крови. Лечение пациентов, пригодных для трансплантации, включает индукцию с помощью бортезомиб-содержащих триплетов (VCD, PAD, RVD) с последующей ВДТ и аутоТГСК и поддерживающей терапией леналидомидом. Обсуждаемыми вопросами остаются длительность поддерживающей терапии, целесообразность консолидации и двойной аутоТГСК для отдельных категорий пациентов. Для пациентов, которых не рассматривают в качестве кандидатов для трансплантации, длительная терапия леналидомидом (Rd) обеспечивает максимальную продолжительность первой ремиссии по сравнению с ограниченной терапией. К альтернативным режимам относятся бортезомиб-содержащие программы VD и VMP.


