Первичная гипероксалурия – редкое аутосомно-рецессивное заболевание, характеризующееся врожденным нарушением метаболизма оксалатов. В настоящее время в мировой литературе описано три типа первичной гипероксалурии. Основным проявлением всех типов заболевания являются повышенная экскреция оксалатов, возвратный уролитиаз и/или нефрокальциноз, прогрессирующее снижение скорости клубочковой фильтрации (СКФ) [1]. Причиной развития нефрокальциноза среди 44 % детей считаются метаболические нарушения различной этиологии, в т. ч. первичная гипероксалурия 1-го типа,
которая наблюдается в 8 % случаев [2].
Первичная гипероксалурия 1-го типа (ОMIM # 259900) обусловлена мутациями в гене аланин-глиоксилат аминотрансферазы (AGXT), кодирующем пероксисомный фермент (AGT), локализующийся в печени [3, 4]. Снижение активности фермента AGT, катализирующего превращение глиоксилата в глицин, приводит к образованию из глиоксилата оксалатов (рис. 1). Оксалаты в свою очередь образуют нерастворимые
соли кальция, накапливающиеся в почках и других органах и тканях. В структуре первичной гипероксалурии на долю 1-го типа приходится около 80 % случаев [5].
Первичная гипероксалурия 2-го типа (ОMIM # 260000) обусловлена мутацией в гене глиоксилат редуктазы/гидроксипируват редуктазы (GRHPR), цитозольного фермента, катализирующего превращение глиоксилата в гликолат и гидроксипирувата в D-глицерат в печени [6–8]. Вследствие мутации в гене GRHPR происходит повышенное образование оксалатов и L-глицерата с последующим образованием солей кальция. На долю первичной гипероксалурии 2-го типа приходится 10 % от общего числа первичной гипероксалурии [5].
Первичная гипероксалурия 3-го типа (ОMIM # 613616) связана с мутациями в гене DHDPSL, кодирующем белок, который преимущественно экспрессируется в печени и почках, по своей структуре сходный с митохондриальным ферментом дигидродипиколинат синтазой или 4-гидрокси-2-оксоглутарат альдолазой. Метаболические реакции, катализируемые данным ферментом, в настоящее время до конца не известны [9]. Составляет около 10 % от общего числа первичной гипероксалурии [5].
Заболеваемость первичной гипероксалурией 1-го типа в Европе составляет 1 : 120 тыс. новорожденных в год [10], распространенность колеблется от 1,05 до 2,9 случая на 1 млн населения во Франции, Швейцарии и Нидерландах [10–12]. В структуре различных причин терминальной
хронической почечной недостаточности (тХПН) у детей первичная гипероксалурия 1-го типа, по данным национальных регистров США, Европы, Японии, составляет менее 1 % [13–15].
При нарушении метаболизма оксалатов почки являются главным органом-мишенью заболевания. При первичной гипероксалурии 1-го типа чрезмерная продукция оксалата приводит к повышенной фильтрации оксалата в клубочках, повышению его концентрации в проксимальных канальцах и
как следствие – отложению кристаллов в интерстиции, а также в собирательной системе почек. Высокий уровень оксалатов оказывает прямое токсическое действие на клетки почечных канальцев и может оказывать токсическое воздействие на другие органы и ткани.
Первые клинические проявления первичной гипероксалурии 1-го типа в 48 % случаев появляются до 4 лет, в 16 % случаев – до 10 и в 20 % случаев – до 20 лет [16]. В зависимости от возраста появления первых клинических проявлений заболевания выделяют инфантильную (4–12 месяцев), ювенильную (после 1 года) и поздноманифестирующую формы (после 25 лет). Инфантильная форма встречается в
10 % случаев первичной гипероксалурии 1-го типа, характеризуется плохой прибавкой в весе, инфекцией мочевой системы, в большинстве случаев диффузным нефрокальцинозом (91 %), реже уролитиазом (44 %), ранним развитием тХПН (в среднем к 3 годам в 80 % случаев), системным
паренхиматозным оксалозом [17, 18]. Ювенильная форма составляет 80–90 % случаев заболевания, проявляется после 1 года болями в животе, гематурией, чаще – симптомами уролитиаза (84 %), реже – нефрокальциноза (41 %), более медленными темпами снижения функции почек [17, 18]. В среднем тХПН при этой форме заболевания достигается в возрасте от 24 до 33 лет [19, 20].
Поздняя манифестация заболевания (менее 10 %) может сопровождаться редким отхождением конкрементов и длительно сохранными почечными функциями.
При развитии ХПН со снижением СКФ ниже 30–50 мл/мин/1,73 м2 уровень плазменного оксалата увеличивается, происходит отложение кристаллов оксалатов кальция (Ох/Са) в различных органах и тканях, за исключением печени. Депозиты Ох/Са определяются в сетчатке глаза, щитовидной
железе, в стенках каротидных артерий с развитием ишемии и изменением функции миокарда. Отложение оксалата в костной ткани сопровождается болями в костях, самопроизвольными
переломами. Рентгенологические признаки оксалатной остеопатии включают плотные метафизарные полосы, расположенные у края метафизов, склероз прилегающих диафизов, кистозные изменения костей, деформации, поднадкостничную резорбцию, увеличение плотности костной ткани в позвонках и гребнях подвздошных костей [21, 22].
Для диагностики первичной гипероксалурии 1-го типа необходимо исследование экскреции оксалатов в пересчете на креатинин мочи (Ox/Cr индекс) (см. таблицу) [23, 24]. С целью дифференцирования первичной и вторичной гипероксалурии, а также различных типов первичной гипероксалурии целесо-
образно исследование экскреции органических кислот, таких как гликолевая кислота, глиоксиловая кислота и L-глицерат. При первичной гипероксалурии 1-го типа в 75 % случаев определяется высокая экскреция гликолевой кислоты, а в 25 % случаев она может быть в пределах нормы, что, возможно,
связано с митохондриальной дислокацией фермента AGT [25]. При отхождении конкрементов необходимо определять их химический состав, который при всех типах первичной гипероксалурии представлен 100 %-ным оксалатом моногидратом (веввелит) [26]. Для определения каталитической
активности фермента AGT проводится биопсия печени, однако и этот метод не всегда позволяет с точностью подтвердить или опровергнуть диагноз первичной гипероксалурии 1-го типа. При диспозиции фермента AGT из пероксисом в митохондрии печени может определяться нормальная активность фермента в биоптатах печени, тогда как субстрат для переаминирования отсутствует. По мере снижения функционального состояния почек отмечается снижение экскреции оксалатов
с нарастанием их концентрации в крови. Так, у больных с СКФ < 30 мл/мин/1,73 м2 концентрация оксалата крови не превышает 20–30 ммоль/л, а у больных с тХПН плазменная концентрация оксалата превышает 50 ммоль/л (норма – 2–9 ммоль/л) [27].
В настоящее время наиболее информативным методом диагностики первичной гипероксалурии 1-го типа является молекулярно-генетическое исследование гена AGXT, локализованного на хромосоме 2q37.3 [4]. AGXT состоит из 11 экзонов и кодирует фермент аланин-глиоксилат аминотрансферазу,
катализирующую переаминирование глиоксилата в глицин, представленного 392 аминокислотами с молекулярной массой около 43 kDa (рис. 1) [28].
К настоящему времени описано 146 мутаций в гене AGXT, ответственных за развитие первичной гипероксалурии 1-го типа, в основном представляющих собой однонуклеотидные замены (75 %), из них 73 миссенс-мутации, 19 нонсенсмутаций, 18 сплайт-мутаций; кроме того, в список включено
36 крупных и малых перестроек гена [29]. Наиболее часто мутации затрагивают 1-ю, 2-ю, 4-ю и 10-ю экзоны гена. Мутации приводят к нарушению активности фермента AGT или формируется нестабильный белок, который подвергается быстрому разрушению.
На биохимическом уровне описаны следующие изменения фермента [30]:
• отсутствие иммунореактивной и каталитической активности фермента AGT (42 %);
• наличие иммунореактивности при полном отсутствии каталитической активности AGT (16 %);
• наличие и иммунореактивной и каталитической активности AGT, которые достигают почти 50 % от нормальных значений. Это связано с дислокацией AGT из пероксисом в митохондрии печени (42 %) [31].
С учетом значительной клинической гетерогенности заболевания с различными темпами прогрессирования в тХПН остается открытым вопрос о возможности выявления клинико-генетических ассоциаций с целью прогнозирования течения заболевания. Выявляемые в родственных браках
“уникальные” мутации не определяются у других больных, а среди большинства пациентов из неродственных браков определяются компаунд гетерозиготные мутации. В некоторых семьях с одинаковым генотипом наблюдается выраженный клинический полиморфизм – от инфантильной формы до относительно бессимптомного течения [32, 33]. В двух ретроспективных исследования, выявивших связь между мутацией p.Gly170Arg (20–40 % от общего числа первичной гипероксалурии 1-го типа) и ответом на терапию пиридоксином, наблюдалось снижение уровня экскреции оксалатов практически до нормального уровня при наличии гомозиготной мутации и частичный эффект в случае гетерозиготной мутации [34,35, 12]. Некоторые мутации встречаются среди определенных этнических групп; наиболее очевидным примером является p.Ile244Thr-мутация (4–9 %), которая определяется у многих пациентов из Северной Африки испанского происхождения [36]. Но и в этих небольших этнических группах установить клинико-генетические ассоциации не удалось. В настоящее время изучается влияние различных факторов и генов-модификаторов на течение заболевания [37].
С целью демонстрации клинико-генетической гетерогенности первичной гипероксалурии 1-го типа приводим клинические наблюдения за двумя пациентами, обследованными в отделении наследственных и приобретенных болезней почек ФГУ “МНИИ педиатрии и детской хирургии” Минздравсоцразвития России с диагнозом “первичная гипероксалурия 1-го типа”.
Таблица. Референтные значения экскреции оксалата, гликолата и L-глицерата с мочой (модификация Zhang X et al., 2003 [55]).
Рисунок 1. Схема преобразования оксалата, гликолата и глиоксилата в гепатоцитах печени. AGX-аланин-глиоксилат аминотрансфераза, GRHPR-глиоксилат редуктаза/гидроксипируват редуктаза, LDH-лактатдегидрогеназа, DAO-D-аминокислот оксидаза [56].
Мальчик С.М. 7 лет. Ребенок от 1-й беременности, протекавшей с токсикозом на всем протяжении, неосложненных родов, из семьи здоровых родителей, не состоявших в родстве. У дедушки и прабабушки по линии отца мочекаменная болезнь. Родился с массой 4160 граммов (75-й перцентиль), длиной 52 см
(75-й перцентиль). До полугода развивался нормально. В возрасте 7 месяцев при диспансерном обследовании было проведено УЗИ почек, выявившее двусторонний медуллярный нефрокальциноз.
С того же возраста появилось ежедневное отхождение мелких конкрементов, периодически сопровождавшееся приступами почечной колики. Ребенок неоднократно госпитализировался в стационары по месту жительства, где выявлялась высокая экскреция оксалатов. Впервые в отделение наследственных и приобретенных болезней почек ФГУ “МНИИ педиатрии и ДХ” Минздравсоцразвития России ребенок поступил в возрасте 3 лет. При поступлении физическое развитие среднее гармоничное (25-й перцентиль по росту и массе), костных деформаций, полиурии, полидипсии не отмечено, отхождение конкрементов ежедневное. Клинический и биохимический анализы, электролитный и
кислотно-основной состав крови в пределах нормы. Экскреция оксалатов была резко повышенной (Ox/Cr = 1,5 при N <0,1 ммоль/ммоль), тогда как экскреция кальция (Ca/Cr = 0,01), уратов (ураты/Cr = 0,2) в пределах нормы; качественная реакция на цистин отрицательная. Аминокислотный спектр крови и мочи
в пределах нормы. При оценке функционального состояния почек наблюдалось снижение концентрационной функции (удельный вес мочи – 1002–1007) при незначительном снижении СКФ
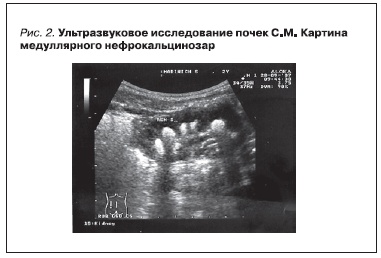
по Шварцу 90 мл/мин/1,73 м2. По данным УЗИ почек выявлен выраженный медуллярный нефрокальциноз при неизмененной эхогенности паренхимы кортикального слоя (рис. 2). На обзорной
рентгенограмме брюшной полости определялись множественные рентген-позитивные конкременты в медуллярном слое почек. При исследовании экскреции органических кислот выявлено повышение уровня гликолевой кислоты (гликолевая кислота/Cr = 318,7 при N = 28–126 ммоль/ммоль). Химический состав
камней представлен 100 %-ным веввелитом (кальция оксалат моногидрат). С учетом стабильно высокого уровня экскреции оксалатов, повышения экскреции гликолевой кислоты, наличия кортико-медуллярного нефрокальциноза и возвратного уролитиаза с раннего возраста у ребенка с отягощенным семейным
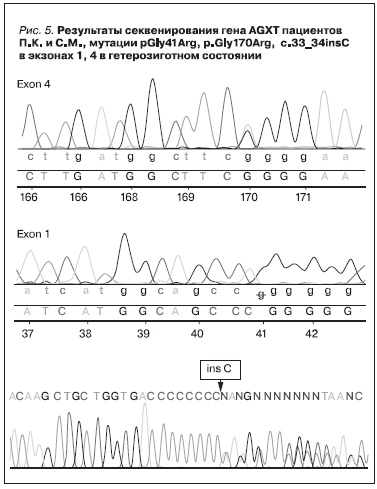
анамнезом по МКБ предположительным диагнозом была первичная гипероксалурия 1-го типа. С целью выявления мутации в гене AGXT проведено молекулярно-генетическое исследование. При селективном скрининге экзонов 1, 4, 10 гена AGXT выявлена мутация в экзоне 1 c.33_34insC в гетерозиготном состоянии (рис. 3), что подтвердило наличие у ребенка первичной гипероксалурии 1-го типа. Второй мутантный аллель в процессе данного исследования не обнаружен.
За двухлетний период наблюдения (2007–2009) на фоне терапии пиридоксином (12 мг/кг/сут) цитратной смесью с соблюдением высокожидкостного режима (> 2 л/1,73 м2/сут, дробно) у ребенка сохранялась стабильно высокая экскреция оксалатов (Ox/Cr = 0,9–1,1 при N = 0,08–0,07 ммоль/ммоль). Функциональное состояние почек оставалось стабильным (СКФ по Шварцу = 80–100 мл/мин/1,73 м2). УЗИ почек выявило повышение выраженности нефрокальциноза с вовлечением и кортикального слоя почек.
У ребенка продолжается пассаж камней с высоким риском развития обструктивной уропатии. Совместно с трансплантологами обсуждается целесообразность проведения изолированной трансплантации печени с целью продления функциональной сохранности почек.
Девочка К.П. 6 лет. Ребенок от первой нормально протекавшей беременности, неосложненных родов, из семьи здоровых, не состоящих в родстве родителей. Родилась на 40-й неделе гестации с массой 2800 граммов (10-й перцентиль), длиной 49 см (25-й перцентиль), до трех лет росла и развивалась нормально.
Семейный анамнез отягощен по материнской линии – у дяди, брата бабушки и брата прабабушки мочекаменная болезнь без нарушения функции почек. Впервые изменения в анализах мочи в виде микрогематурии и абактериальной лейкоцитурии выявлены при диспансерном обследовании в возрасте 3 лет, однако углубленного обследования не проводилось, анализы мочи нормализовались на фоне уросептической терапии. В возрасте 4,5 лет при проведении УЗИ почек выявлен медуллярный нефрокальциноз. В отделении наследственных и приобретенных болезней почек ФГУ “МНИИ педиатрии и ДХ” Минздравсоцразвития России ребенок наблюдается с 4 лет 9 месяцев. При поступлении физическое развитие среднее гармоничное (50-й перцентиль по росту и массе), костных деформаций, полиурии, полидипсии не отмечено. Клинический и биохимический анализы крови, электролиты и кислотно-основное состояние крови в пределах нормы. Уровень паратгормона, остекальцина соответствовал
возрастной норме. Экскреция кальция (Са/Сr = 0,08), уратов (ураты/Cr = 0,17) не нарушена, реабсорбция фосфатов не снижена (93 %), экскреция оксалатов непостоянно повышена (Ox/Cr = 0,08–0,25). Качественная реакция на цистин отрицательная. Аминокислотный состав крови и мочи был в пределах
возрастной нормы. При исследовании экскреции органических кислот выявлено повышение глицериновой кислоты до 15,9 ммоль/ммоль Сr (норма – 0–8) при нормальном уровне гликолевой и
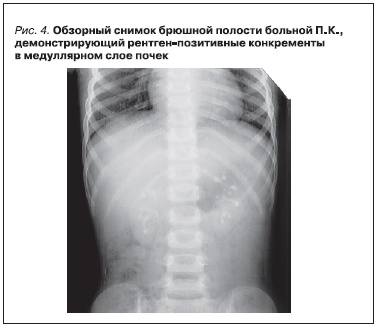
глиоксиловой кислот. УЗИ подтвердило наличие медуллярного нефрокальциноза при нормальных возрастных размерах почек (50-й перцентиль). При проведении экскреторной урографии выявлены рентген-позитивные конкременты в медуллярном слое паренхимы почек (рис. 4). СКФ по Шварцу 90 мл/мин/1,73 м2, концентрационная функция сохранна. С учетом повышенного уровня экскреции глицериновой кислоты у ребенка с медуллярным нефрокальцинозом без уролитиаза предполагалось наличие первичной гипероксалурии 2-го типа, в связи с чем проводилось молекулярно-генетическое исследование всех экзонов гена GRHPR – мутации выявлено не было. При исследовании всех кодирующих экзонов гена AGXT выявлено две мутации в гетерозиготном состоянии: в экзоне 1 c.121G > A (pGly41Arg), в экзоне 4 c.508G > A (р.Gly170Arg), что подтвердило наличие у ребенка первичной гипероксалурии 1-го типа (рис. 5). На фоне постоянного приема пиридоксина в дозе 13 мг/кг/сут, препаратов магния и цитратной смеси в течение года наблюдалась нормализация экскреции оксалатов (Ox/Cr = 0,04–0,06), однако продолжалось прогрессирующее снижение фильтрационной функции почек (СКФ по Шварцу 60 мл/мин/1,73 м2). С учетом быстрых темпов прогрессирования заболевания ребенок был консультирован трансплантологами с целью определения сроков возможной изолированной трансплантации печени. В настоящее время проводится обследование родителей для определения потенциальных доноров для родственной трансплантации печени.
Таким образом, в первом случае у ребенка 7 лет с первичной гипероксалурией 1-го типа, инфантильной пиридоксинрезистентной формой с нефрокальцинозом и возвратным уролитиазом наблюдается длительная сохранность функционального состояния почек, тогда как, по данным литературы, при инфантильной форме заболевания тХПН наступает в 80 % случаев до 3 лет. Во втором случае у девочки с ювенильной пиридоксин-чувствительной формой заболевания с изолированным нефрокальцинозом отмечается прогрессирующее снижение функции почек. Выявленные у второго ребенка мутации в гене AGXT (pGly41Arg, р.Gly170Arg) относят к т. н.
дислокационным мутациям (приводят к “ошибочному” переносу фермента из пероксисом в митохондрии гепетоцитов) с хорошим ответом на терапию высокими дозами пиридоксина. Положительный ответ на пиридоксин проявляется снижением уровня экскреции оксалатов с замедлением темпов прогрессирования заболевания, однако у девочки на терапии пиридоксином при снижении экскреции оксалатов до нормального уровня продолжалось снижение СКФ. Полученные данные еще раз свидетельствуют о сложности проведения клинико-генетических ассоциаций при первичной гипероксалурии 1-го типа.
Всем больным с первичной гипероксалурией независимо от типа рекомендуется повышенный питьевой режим (от 2 до 3 л/м2), что подтверждается крупным эпидемиологическим и проспективным исследованием о влиянии потребления большого количества жидкости для предотвращения камнеобразования [38]. Жидкость необходимо потреблять круглосуточно, без длительных безводных промежутков, для чего детям раннего возраста устанавливают гастростому для введения необходимого объема жидкости. Диетические мероприятия по ограничению поступления с пищей оксалатов и кальция при данной патологии нецелесообразны, т. к. не предотвращают прогрессирования заболевания, а лишь ухудшают качество жизни пациента. Патогенетически обосновано использование высоких доз (10–30 мг/кг) пиридоксина при первичной гипероксалурии 1-го типа, т. к. он является кофактором AGX, однако положительный эффект от терапии пиридоксином в настоящее время выявлен у пациентов с определенными мутациями в гене AGXT (p.Gly170Arg, p.Phe152Ile). Положительным ответом на терапию пиридоксином считается снижение экскреции оксалатов минимум на 30 % от исходного уровня при использовании максимальной дозы в течение 1–3
месяцев [39]. С целью ингибирования оксалатно/кальциевой кристаллизации рекомендуется использование цитратов, в частности цитрата калия в дозе 100–150 мг/кг/сут, которые образуют нерастворимые соединения с кальцием, выводя его из организма [40]. Необходим постоянный прием препаратов фосфора (ортофосфат 30–40 мг/кг/сут) и магния (оксид магния 300–500 мг/сут/м2).
В настоящее время продолжается оценка эффективности использования пробиотиков – Oxalobacter formigenes (облигатные анаэробные бактерии, естественно колонизирующие желудочно-кишечный тракт), который метаболизирует оксалаты в кишечнике, препятствуя их всасыванию [37, 41].
При достижении тХПН пациентам с первичной гипероксалурией 1-го типа для профилактики развития системного оксалоза рекомендуется проведение ежедневного гемодиализа высокого потока в сочетании с перитонеальным диализом или длительных ежедневных сеансов гемодиализа [42, 43].
В настоящее время, по данным различных авторов, на момент установления диагноза первичной гипероксалурии 1-го типа высокая доля больных (10–40 %) уже достигают тХПН [5, 10, 11, 19, 44]. Выбор варианта трансплантации (изолированная трансплантация печени или почек, комбинированная трансплантация печени и почек) проводится в зависимости от степени снижения функционального состояния почек (СКФ) [45]. Для оптимизации трансплантационной стратегии решение вопроса о сроках и виде трансплантации органов необходимо планировать до достижения пациентами тХПН.
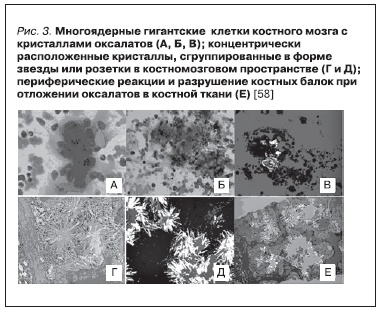
Изолированная трансплантация почки, впервые выполненная больному с первичной гипероксалурией 1-го типа в 1969 г., в настоящее время считается малоэффективным методом. Связано это с высоким процентом возврата нефрокальциноза и уролитиаза в трансплантате в короткие сроки после
трансплантации и как следствие – низкой выживаемостью почечного трансплантата и плохим качеством жизни больных. Так, по данным регистра Европейской ассоциации диализа и трансплантации (ERA-EDTA), 3-летняя выживаемость трансплантата почки среди реципиентов с первичной гипероксалурией 1-го типа составляет 23 % от живого и 17 % от трупного донора [46]. Кроме того, изолированная трансплантация почки не предотвращает прогрессирования системного
оксалоза с вовлечением костной и сосудистой патологии [10, 47]. Частичная успешность изолированной трансплантации почки может быть достигнутой при наличии остаточной активности фермента AGT и сохранении остаточной функции почки на момент трансплантации (СКФ – 20–30 мл/мин/1,73 м2) с проведением диализа в послеоперационный период. В случае доказанной пиридоксин-чувствительности при изолированной трансплантации почки необходимо продолжать
прием пиридоксина с целью снижения экскреции оксалатов и предотвращения рецидива нефрокальциноза и уролитиаза в трансплантате. Проведение пред- и посттрансплантационного диализа направлено на снижение плазменного Ох/Са и может снижать риск повторного осаждения оксалатов в трансплантате [48].
При СКФ в диапазоне 50–70 мл/мин/1,73 м2 предлагается проведение изолированной трансплантации печени, позволяющей полностью восстанавливать активность фермента AGT со стабилизацией или улучшением функционального состояния почек, возможным растворением отложений Ох/Са в медуллярном /кортикальном слоях почек и предотвращением развития системных осложнений [49]. Однако сохраняется этическая проблема, связанная с удалением “здоровой” по всем остальным параметрам печени [50, 51].
Комбинированная трансплантация печени и почек у больных с первичной гипероксалурией 1-го типа целесообразна при СКФ в пределах 40–20 мл/мин/1,73 м2, т. к. значительно повышается интенсивность отложения Ох/Са во всех органах и тканях, а функциональное состояние почек продолжает ухудшаться [46]. В этой ситуации трансплантация печени заменяет фермент-дефицитный орган, а трансплантация почки служит для замены функции пораженного основного органа-мишени. Кроме того, существует теория о том, что комбинированная гепаторенальная трансплантация имеет иммунологическое преимущество; так, своевременное лечение острого отторжения в одном органе может предотвратить отторжение другого органа. К тому же предполагается, что лимфотоксические
антитела могут исчезать после пересадки печени, возможно за счет абсорбции их непаренхимальными клетками печени [52]. По данным Европейского и Американского регистров по первичной гипероксалурии 1-го типа, комбинированная трансплантация печени и почек у взрослых больных имеет лучшие результаты выживаемости трансплантата: 1-, 2- и 5-летняя выживаемость пациентов составляет 88, 80 и 72 % соответственно, а выживаемость трансплантата – 82, 78 и 62 % соответственно [18, 50]. Сравнительный анализ различных вариантов трансплантации почек и печени у 58 взрослых больных с первичной гипероксалурией 1-го типа показал лучшую 3-летнюю выживаемость трансплантата при комбинированной трансплантации печени и почек (95 %) по сравнению с изолированной трансплантацией почки (56 %) [50]. При этом следует отметить, что в детском возрасте, особенно в группе до 5 лет, смертность после комбинированной трансплантации печени и почек может достигать 40 %, хотя уже в следующей возрастной группе (5–10 лет) выживаемость пациентов составляет около 80 % [53, 54]. Комбинированная трансплантация печени и
почек может выполняться последовательно или одновременно.
После комбинированной трансплантации печени и почек происходит нормализация эндогенной продукции оксалатов, однако экскреция оксалатов с мочой может оставаться значительно повышенной. Это связано с длительной резорбцией системных отложений Ох/Са и может служить риском для развития нефрокальциноза и уролитиаза в почечном трансплантате. В связи с этим как при комбинированной гепаторенальной трансплантации, так при изолированной трансплантации
печени и почек необходимо продолжать повышенное потребление жидкости (до 3 л/м2) и использование ингибиторов кристаллизации в первые 6–12 месяцев после трансплантации. Однозначного мнения по поводу проведения ежедневного гемодиализа после трансплантации не достигнуто; с одной стороны, возможно быстрое удаление плазменного оксалата кальция, связанного с резорбцией системных отложений, с другой – велик риск перенасыщения Ox/Ca за счет уменьшения объема мочи.
Таким образом, представленные клинические наблюдения пациентов с первичной гипероксалурией 1-го типа подчеркивают необходимость ранней диагностики заболевания с целью решения вопроса о виде и сроках проведения трансплантации органов. Консолидированное сотрудничество детских, взрослых нефрологов и трансплантологов может способствовать ранней диагностике первичной гипероксалурии с оптимизацией консервативного лечения и определением четкой трансплантационной стратегии для увеличения выживаемости трансплантатов и больных с улучшением качества их жизни.
