
Введение
Идиопатический нефротический синдром (НС) характеризуется тяжелой протеинурией, гипоальбуминурией, отеками и гиперлипидемией и возникает главным образом у детей. По отношению к стероидной терапии НС делится на стероидчувствительный и стеродрезистентный (СРНС). В детском возрасте большинство пациентов отвечают на глюкокортикоидную терапию, однако для 10—20 % больных отмечается стероидная резистентность, что является причиной прогрессирования заболевания с формированием почечной недостаточности [1, 2]. Накопленные данные о патогенезе идиопатического НС позволяют предположить, что часть детей с СРНС могут иметь первичный дефект барьера клубочкового фильтрата [3-6]. Недавние исследования показали, что мутации в гене NPHS2, кодирующего подоцин (структурный белок подоцита), ответственны за развитие аутосомно-рецессивного НС[4], но также могут быть причиной формирования спорадически возникшего СРНС, наблюдаемого, по данным разных авторов, в 10,5-28 % [1, 2, 7, 8]. Однако в других случаях в основе патогенеза СРНС лежат иммунологические сдвиги с преимущественным дисбалансом Т-лимфоцитов. Т-клетки у больных СРНС секретируют циркулирующий фактор, который повреждает клубочковый фильтрационный барьер [6]. В последнее время были предприняты усилия по изучению фармакокинетики глюкокортикоидов (ГК) у детей с почечной патологией, их взаимодействию с глюкокортикоидными рецепторами (ГР) и аффинности последних [9-11]. Молекулярные механизмы, лежащие в основе стероидной резистентности в спорадических формах НС, обусловленного дисбалансом Т-лимфоциты, до сих пор не выявлены.
Влияние на клетки глюкокортикоиды оказывают через глюкокортикоидный рецептор. Рецепторы связываются с ГРЭ-элементами, расположенными в промоторных областях генов-мишеней и регулируют их транскрипционную активность. Также рецепторы взаимодействуют с другими транскрипционными факторами посредством белок-белок взаимодействий и взаимно репрессируют или стимулируют друг друга в транскрипционной активности других генов.
Белок глюкокортикоидного рецептора состоит из нескольких доменов. ДНК-связывающий домен расположен в центральной части молекулы глюкокортикоидного рецептора. C-конец глюкокортикоидного рецептора является гормон-связывающим доменом. Внутри этого домена расположен также трансактивирующий район тау-2 [12] .
Район тау-1 расположен в N-концевом (иммуногеном) домене глюкокортикоидного рецептора. При этом район тау-1 обеспечивает 60-70 % активности этого белка. Именно его избыток блокирует транскрипцию с минимального CYO-промотора in vivo и in vitro [13].
Иммуногенный домен глюкокортикоидного рецептора кодируется одним большим экзоном, каждый из цинковых пальцев - отдельным экзоном, и пять экзонов кодируют гормон-связывающий домен [14].
Мутации в гене глюкокортикоидного рецептора приводят к развитию спорадического синдрома глюкокортикоидной резистентности (sporadic glucocorticoid resistance syndrome). А для полиморфного маркера rs41423247 (BclI)показана ассоциация с повышенным весом. Этот полиморфный маркер находится в 400-м положении в матричной РНК и представляет собой замену С/G, гомозиготный G аллель ведет к высокому уровню инсулина и большей инсулин-резистентности по сравнению с группой, содержащей генотип CC или GC. Кроме того, показана ассоциация между наличием этого маркера у пациентов и повышенной брюшной тучностью в среднем возрасте [15].
Полиморфизм rs6195 (N363S) идентифицирован в 363-м кодоне во 2-м экзоне, этот полиморфный маркер выглядит как изменение ААТ на AGT, в результате в белке происходит замена аспарагина на серин. Была описана связь данного полиморфизма с глюкокортикоидной гиперчувствительностью [16].
Два полиморфных маркера, rs6189 и rs6190 (ER22/23EK), жестко сцеплены между собой и представляют собой две мутации в положениях 22 и 23 (2-й экзон). Первая мутация незначима, поскольку не ведет к аминокислотной замене (замена GAG на GAA), вторая же мутация в 23-м кодоне представляет собой замену AGG на AAG и ведет к замене аргинина на лизин. По некоторым данным, при наличии этого полиморфизма у пациентов в крови присутствует более низкая концентрация холестерина и С-реактивного белка, а также отмечается снижение эффективности дексаметазона [17]. Ассоциация полиморфного маркера rs10052957 (Tth111I) с высоким уровнем кортизола показана в популяции мужчин-шведов. При наличии этой мутации происходит замена аденина на гуанин в 256-м положении мРНК.
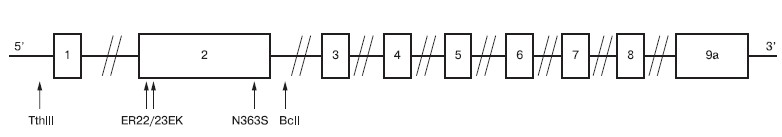
В связи с тем что стероидные препараты широко используются для лечения НС, нами была исследована ассоциация полиморфных маркеров rs10052957, rs41423247, rs6195 (rs57667111), rs6189, rs6190 в гене глюкокортикоидного рецептора с резистентностью к глюкокортикоидной терапии (см. рисунок).
Рисунок. Позиционирование полиморфизмов гена глюкокортикоидного рецептора у детей с СРНС.
Материал и методы
Под нашим наблюдением находились 58 (35 девочек, 23 мальчика) детей в возрасте от 5 месяцев до 18 лет, средний возраст - 11,8 ± 5,45 года. Всем детям был диагностирован СРНС на основании стандартного обследования, в которое входило определение сывороточного белка, альбумина, холестерина, триглицеридов, креатинина, а также исследование мочи. Стероидрезистентность была диагностирована в случае отсутствия ответа на терапию при приеме преднизолона в дозе 2 мг/кг/сут в течение 6-8 недель. Всем детям проведена нефробиопсия с определением морфологической формы. Были выявлены следующие морфологические формы: фокально-сегментарный гломерулосклероз (29 детей), минимальные изменения (9), IgM-нефропатия (6), IgA-нефропатия (5), мембрано-пролиферативный (5), мезангиопролиферативный (3) и 1 ребенок с фибропластическим гломерулонефритом. Все дети находились на стационарном обследовании и лечении в Российской детской клинической больнице в отделении нефрологии с 2002 по 2011 г.
Контрольная группа представлена 40 пациентами в возрасте 27-73 лет без воспалительных и эндокринных заболеваний.
В работе использовали термостабильную ДНК-полимеразу Taq, полученную от фирмы “Fermantas” (Вильнюс, Литва). Олигонуклеотидные праймеры синтезированы ЗАО “Синтол” (Москва). Геномную ДНК выделяли из цельной крови больных посредством экстракции фенолом-хлороформом после инкубации образцов крови с протеиназой К в присутствии 0,1 %-ного додецилсульфата натрия [18].
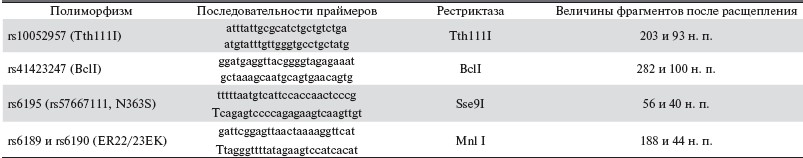
Амплификация полиморфного участка гена проведена с помощью полимеразной цепной реакции (ПЦР) на термоциклере “Терцик” (ЗАО “ДНК- Технология”, Москва) в 25 мкл реакционной смеси следующего состава: 70 мМ Трис-HCl, pH = 8,8, 16,6 мМ сульфат аммония, 0,01%-ный Твин-20, 2 мМ хлорида магния, 0,2 мМ dNTP, по 33 нг праймеров, 1,5 ЕД полимеразы Taq, 50-100 нг геномной ДНК. Условия амплификации фрагмента ДНК: 94 °C/2 мин - 1-й цикл; 95 °C/30 с, 60 °C/30 с (62 °C/30 с), 72 °C/20 с - 35 циклов; 72 °C/6 мин - последний цикл. Размеры продуктов амплификации для полиморфных маркеров были следующими: 296 п. о. для rs10052957, 382 п. о. для rs41423247, 261 п. о. для rs6195 (rs57667111), 232 п. о. для rs6189 и rs6190 (табл. 1).
Таблица 1. Праймеры и рестриктазы, используемые в работе.
Продукты рестрикции анализировали с помощью электрофореза в 8%-ном полиакриламидном геле и в 2%-ном агарозном геле с последующей окраской бромистым этидием. В качестве маркера молекулярной массы использованы ДНК плазмиды pUC19, расщепленной рестриктазой MspI.
Статистическая обработка данных проведена при помощи компьютерной программы для статистического анализа Statistica 7,0. Для проверки статистической значимости различий частотных показателей использовали критерий χ2 по Пирсону. При количестве наблюдений меньше 5 использован точный критерий Фишера. Достоверными считались различия при p < 0,05; 0,05 ≤p < 0,1 рассматривали как тенденцию к различию.
Для описания относительного риска развития заболевания рассчитывали отношение шансов (OR). Вычисления производили с помощью программы Calculator for confidence intervals of odds ratio (David Hutchon). OR = 1 рассматривали как отсутствие ассоциации; OR > 1 - как положительную ассоциацию (“повышенный риск развития патологии”), OR < 1 - как отрицательную ассоциацию аллеля или генотипа с заболеванием (“пониженный риск развития патологии”). Также рассчитывали величину доверительного интервала (CI, confidence interval) - интервала значений, в пределах которого с вероятностью 95 % находится ожидаемое значение рассматриваемого параметра, в данном случае значение OR.
Результаты и обсуждение
Человеческий ген ГР NR3C1 располагается на хромосоме 5q31.3 и состоит из девяти экзонов [19]. Полиморфизм, т. е. изменения в последовательности ДНК с частотой более 1 % в здоровой популяции в человеческом гене ГР, может приводить к нарушению формирования комплекса ГК-ГР, а затем изменять трансактивные и/или трансподавляющие процессы.
В настоящее время обнаружено множество полиморфизмов, однако лишь немногие из них функциональные. Выявлены ассоциации полиморфных маркеров IthIIII (rs10052957), ER22/23EK (rs6189/rs6190), N363S (rs6195), BclI (rs41423247) и GR-9β (rs6198) с различными метаболическими параметрами и массой тела, аутоиммунными и кардиоваскулярными заболеваниями. Эти генетические полиморфные маркеры ассоциированы с чувствительностью к ГК и изменением уровня кортизола [20] и могут объяснить различия эффективности ГК терапии.
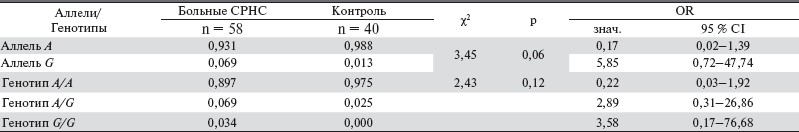
В нескольких исследованиях была изучена ассоциация этих полиморфных маркеров с воспалительными заболеваниями кишечника, в которых не было выявлено ассоциации между полиморфным маркером ER22/23EK и эффективностью ГК [21]. Три полиморфных маркера изучены на больных с рассеянным склерозом TthIIII, ER22/23EKи 9β-G, гаплотипы которых были ассоциированы с резистентностью к ГК и более быстрым прогрессированием заболевания. Однако именно наличие полиморфизма ER22/23EK, а не двух других полиморфизмов повлияло на полученный результат [21]. В нашем исследовании мы получили высокую ассоциацию стероидной резистентности при НС у детей с аллелем А как в гомозиготном, так и в гетерозиготном состоянии полиморфного маркера ER22/23EK (χ2 = 10,4; p = 0,001, и χ2 = 9,66; p = 0,002) (табл. 2), в то время как распределение полиморфного маркера TthIIII практически соответствовало таковой в контрольной группе (табл. 3).
Таблица 2. Распределение частот аллелей и генотипов полиморфного ER22/23EK гена NR3C1 по сравнению с контрольной группой.
Таблица 3.Распределение частот аллелей и генотипов полиморфного TthIIIIгена NR3C1 по сравнению с контрольной группой.
Проведено лишь несколько исследований роли полиморфизма N363S в чувствительности на экзогенные ГК. V. Szabo et al. (2007) [22] продемонстрировали варианты полиморфных маркеров NR3C1-гена, способствующие развитию стероидной гипертензии. У 102 пациентов, перенесших фоторефракционную кератэктомию и получавших стероиды в рамках послеоперационной терапии, отмечена значительная корреляция между N363S-гетерозиготным генотипом и глазной гипертензией. Кроме того, 48 пациентов с мышечной дистрофией Дюшенна, получавших преднизолон или дефлазакорт, N363S-носители GG-генотипа дольше сохраняли мышечную функцию [23]. Однако в исследовании ассоциации данного полиморфного маркера с воспалительными заболеваниями кишечника не выявлено никаких ассоциаций [24]. Аналогичные данные получены нами. Распределение частот аллелей и генотипов данного полиморфного маркера N363S не отличалось от контрольной группы (табл. 4).
Таблица 4.Распределение частот аллелей и генотипов полиморфного N363Sгена NR3C1 по сравнению с контрольной группой.
Изученная ассоциация полиморфного маркера BclI гена NR3C1 у больных с воспалительными заболеваниями кишечника продемонстрировала, что стероидзависимая форма чаще наблюдалась у детей с СС-генотипом, тогда как пациенты с аллелем G в гетерозиготном и гомозиготном состоянии сохраняли длительную ремиссию [25]. Отсутствие значимой разницы в распределении аллелей и генотипов полиморфного маркера BclI гена NR3C1 с контрольной группой лишь подтверждают роль данного маркера в чувствительности к ГК (табл. 5).
Таблица 5. Распределение частот аллелей и генотипов полиморфного Bcll гена NR3C1 по сравнению с контрольной группой.
Таким образом, обнаруженные нами результаты позволяют делать вывод, что наличие аллели А в полиморфном маркере ER22/23EK у детей со стероидрезистентным НС может обусловливать развитие резистентности к глюкокортикоидной терапии. Нами не выявлено достоверных ассоциаций СРНС с другими полиморфными (Tth111I, N363S, BclI) гена NR3C1.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}