
Введение
Тромботическая микроангиопатия (ТМА) – этиологически гетерогенный клинико-морфологический синдром, характеризующийся поражением сосудов микроциркуляторного русла.
Гистологически ТМА – это особый тип повреждения микрососудов, представленный отеком эндотелиальных клеток с их отслойкой от базальной мембраны, расширением субэндотелиального пространства с накоплением в нем аморфного мембраноподобного материала с образованием тромбов, содержащих тромбоциты и фибрин, что приводит к окклюзии просвета сосуда, вызывая развитие ишемии органов и тканей [1].
ТМА проявляется тромбоцитопенией, возникающей вследствие усиленного потребления тромбоцитов при образовании множественных микротромбов, микроангиопатической гемолитической Кумбс-негативной анемией (механический гемолиз), лихорадкой и в зависимости от вовлечения того или иного участка сосудистого русла – почечной недостаточностью, кардиальными осложнениями, дыхательной недостаточностью, нарушениями зрения, панкреатитом, ишемией кишечника [1]. До появления эффективных методов лечения смертность была высокой, достигая 72–94%.
ТМА классифицируют на первичные и вторичные. Первичные ТМА включают тромботическую тромбоцитопеническую пурпуру, типичный и атипичный гемолитико-уремический синдром (ГУС).
Первичные ТМА
Тромботическая тромбоцитопеническая пурпура (ТТП)
В основе патогенеза ТТП лежит дефицит металлопротеиназы ADAMTS13 (а disintegrin-like and metalloprotease with thrombospondin type I motif 13), которая в норме расщепляет образуемые эндотелием крупные мультимеры фактора Виллебранда (VWF – von Willebrand factor). Дефицит этого фермента приводит к циркуляции в плазме крови пациентов сверхкрупных мультимеров VWF с образованием в системе микроциркуляции тромбов. Дефицит ADAMTS13 может быть обусловлен мутациями в гене ADAMTS13 при редких врожденных формах ТТП (синдром Upshaw–Schulman) или появлением в циркуляции аутоантител к ADAMTS13, являющихся его ингибиторами, что имеет место при более часто встречаемых приобретенных формах ТТП.
Отличительные особенности ТТП:
- активность ADAMTS13 <5–10% (активность ADAMTS13 в норме составляет 80–110%);
- преимущественно экстраренальные проявления, особенно поражение нервной системы (тяжелое поражение почек более свойственно ГУС);
- тяжелая тромбоцитопения (с числом тромбоцитов часто менее 30 тыс. в мм3 при острых эпизодах) [2, 3].
Главная составляющая лечения ТТП – сеансы истощающего плазмообмена. При выявлении ингибиторных антител к ADAMTS13 – добавление к терапии глюкокортикоидов. При неэффективности проводимого лечения может быть использована иммуносупрессивная терапия ритуксимабом (off-label) [3].
Типичный гемолитико-уремический синдром (ГУС)
Типичная (пост-диарейный, STEC – Shiga Toxin-Producing Escherichia Coli) ТМА, опосредованная повреждающим эндотелий действием шига-токсина кишечной палочки (STEC – шига-токсин продуцирующий штамм E. сoli, обычно O157:H7 или O104:H4 серотипов), с преимущественным вовлечением почек – развитием острого повреждения почек (ОПП).
Скрининг на STEC-ГУС необходим всем больным с признаками поражения желудочно-кишечного тракта, особенно с диареей. Лабораторные исследования следует выполнять в первые сутки госпитализации больного до начала антибактериальной терапии. Для диагностики STEC-ГУС показаны посев кала для выявления культуры STEC, определение шига-токсина в кале и сыворотке крови. В отношении лечения рекомендуются адекватная антибактериальная терапия (предпочтительны бактериостатические антибиотики), коррекция водно-электролитных нарушений и при необходимости своевременное начало диализных методов лечения [1].
Атипичный гемолитико-уремический синдром (аГУС)
аГУС – заболевание, опосредованное дисфункцией системы регуляции комплемента с неконтролируемой активацией его альтернативного пути. аГУС чаще всего имеет в основе генные мутации белков – регуляторов системы комплемента: CFH (фактор H) у 20–25% пациентов, MCP (мембранный кофакторный протеин) – у ≈15% и CFI (фактор I) – у ≈10%. Мутации фактора В (CFB) встречаются крайне редко (1%), в то время как мутации C3 фракции комплемента встречаются среди 10% пациентов. Редкими являются мутации гена тромбомодулина (THBD). Терапия включает сеансы плазмообмена и введение экулизумаба (гуманизированного моноклонального антитела к C5 фракции терминальной стадии каскада комплемента).
Диагноз аГУС – это диагноз исключения. Он устанавливается на основании характерной клинической картины и должен быть подтвержден лабораторными данными, исключающими другие причины ТМА.
 В данном обзоре мы более подробно остановимся на вторичных ТМА.
В данном обзоре мы более подробно остановимся на вторичных ТМА.
Вторичные ТМА
ТМА определяют как вторичные, когда их развитие ассоциировано с различными заболеваниями или состояниями. Наиболее частые причины вторичных ТМА – беременность, аутоиммунные заболевания, злокачественные новообразования, прием некоторых лекарственных препаратов, инфекции, трансплантация сóлидных органов или костного мозга (табл. 1).
Клинические проявления вторичных ТМА напрямую зависят от вызвавшей ее патологии. Необходимо четко разграничивать данные состояния, поскольку своевременная диагностика и адекватная терапия существенно улучшают прогноз.
 Злокачественная артериальная гипертензия (ЗАГ)
Злокачественная артериальная гипертензия (ЗАГ)
ЗАГ – ургентное жизнеугрожающее состояние, клинически проявляющееся очень высокими цифрами АД (>180/120 мм рт.ст.) в сочетании с ишемическим поражением органов-мишеней (сетчатка, почки, сердце, головной мозг) вследствие фибриноидного некроза сосудистой стенки [4]. Клинические проявления данного состояния представлены в табл. 2 [5, 6].
По данным литературы, признаки микроангиопатической гемолитической анемии (МАГА) и тромбоцитопении встречаются лишь у 27–44% пациентов с ЗАГ. Патофизиологические механизмы, лежащие в основе ЗАГ, не до конца ясны. Van den Born et al. [7] обнаружили, что активность ADAMTS13 была умеренно снижена (до 80%) у пациентов с тяжелой или злокачественной гипертензией. Они также продемонстрировали, что у пациентов с ТМА, ассоциированной с ЗАГ, уровень фактора фон Виллебранда был выше, чем у пациентов с артериальной гипертензией (АГ) без сопутствующей ТМА. Авторы заключили, что сниженная активность ADAMTS13 и повышенный уровень фактора фон Виллебранда (скорее всего связанный с эндотелиальным повреждением) служат результатом агрегации тромбоцитов и микрососудистым тромбозом [8].
Таким образом, определение активности ADAMTS13 может стать подспорьем для дифференциальной диагностики между ТМА, ассоциированной с ЗАГ, и тяжелой АГ без ТМА еще до получения результатов нефробиопсии [8].
Сложной задачей представляется определение ЗАГ как инициального фактора развития вторичной ТМА или же АГ в структуре первичной ТМА. Снижение активности ADAMTS13 у пациентов с ЗАГ и картиной ТМА в биоптате почки в отсутствие МАГА и тромбоцитопении может указывать на то, что первична именно ЗАГ.
Диагноз «ТМА, ассоциированная с ЗАГ» может быть установлен после исключения первичных ТМА (аГУС, ТТП) или же катастрофического антифосфолипидного синдрома (КАФС), других вторичных ТМА при выявлении гистологической картины ТМА в биоптате почки независимо от наличия или отсутствия е гематологических признаков.
Лечение. Адекватный контроль артериального давления часто улучшает течение ТМА, а отсутствие эффекта от проводимой антигипертензивной терапии должно наводить на мысль о возможном варианте хронического течения аГУС у пациентов с генетическим дефектом регуляции альтернативного пути активации комплемента, в пользу чего свидетельствуют маркеры его активации у пациентов с ЗАГ [9]. Таким образом, ЗАГ в этом случае служит триггером для развития клинических проявлений аГУС, поэтому применение комплемент-блокирующей терапии, по-видимому, может быть оправданно в отношении некоторых пациентов с резистентной ЗАГ и быстронарастающей почечной недостаточностью даже в случаях «неполной» ТМА [10].
Генетическая тромбофилия, вероятно, служит дополнительным фактором риска развития ТМА при ЗАГ, а также вносит свой вклад в выраженность органного поражения у таких пациентов [11].
Злокачественные опухоли
Диагноз ТМА основывается на выявлении МАГА и тромбоцитопении, однако указанные состояния часто возникают как результат противоопухолевого лечения и не связаны с наличием ТМА, а обусловлены гематоксичностью проводимой химиотерапии. Тем не менее ТМА следует предполагать при наличии шизоцитов в периферической крови и отрицательной реакции Кумбса.
Таким образом, ТМА может возникать как при наличии злокачественного новообразования, так и в результате противоопухолевой терапии. Наиболее часто встречаются случаи ТМА, ассоциированные с опухолями сóлидных органов [12]. Тем не менее онкогематологические причины, такие как лимфома, все же занимают около 8% всех случаев. В структуре злокачественных новообразований сóлидных органов, наиболее часто осложняющихся ТМА, – опухоли желудка, легкого, молочной железы, простаты, яичников, а также первичная аденокарцинома (табл. 3) [12].
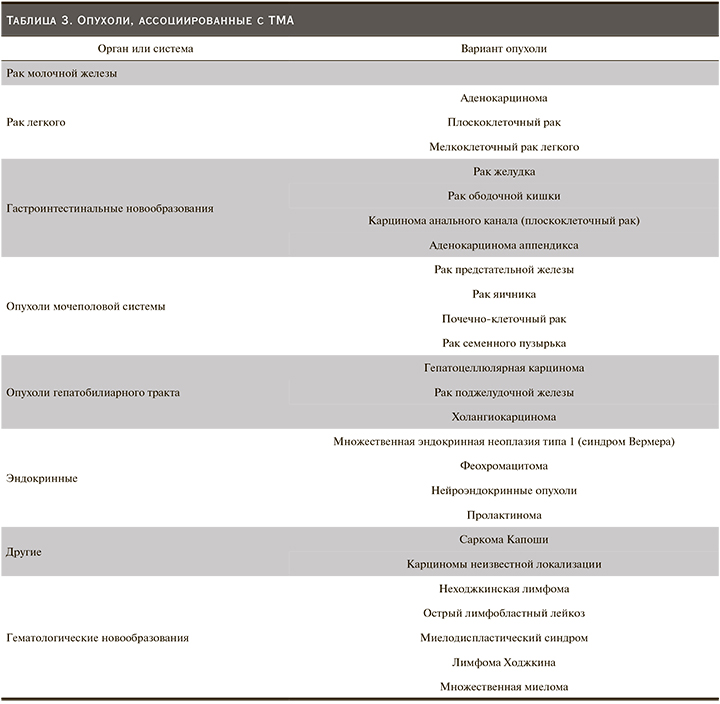
Успешное противоопухолевое лечение в данном случае улучшает прогноз, тогда как плазмотерапия обычно неэффективна [12].
Существуют определенные особенности, позволяющие предположить: возникла ли ТМА в результате наличия злокачественного новообразования, или же это результат применения противоопухолевых препаратов?
Во-первых, метастатическое поражение более характерно для ТМА, ассоциированной со злокачественным новообразованием (более 90% пациентов), в то время как при лекарственно-ассоциированной ТМА признаки активности опухоли зачастую отсутствуют. ТМА часто выявляется у пациентов с метастазами в красный костный мозг.
Во-вторых, опухоль-ассоциированная ТМА часто клинически напоминает ТТП, в то время как пациенты с ТМА, ассоциированной с противоопухолевым лечением, имеют широкий спектр клинических проявлений.
В-третьих, синдром диссеминированного внутрисосудистого свертывания более характерен для ТМА, ассоциированной со злокачественными новообразованиями, чем с ТМА, вызванной противоопухолевыми препаратами.
ВИЧ-ассоциированная ТМА
Развитие ВИЧ-ТМА может быть опосредовано прямым воздействием вируса, косвенным влиянием секретируемых цитокинов или действием протеинов ВИЧ (Tat, gp120, р24) на эндотелиальные клетки [13]. Роль фактора фон Виллебранда не определена из-за недостатка данных об уровне протеаз при ВИЧ-ТМА, описанных в литературе. ВИЧ-ТМА может развиваться как на ранних, так и на поздних стадиях заболевания. ВИЧ-ТМА отличает неблагоприятный прогноз, особенно при полиорганном поражении, с развитием необратимой утраты почечной функции и летальным исходом [14]. В то же время возможны ремиссии заболевания – спонтанные или после применения комбинированной терапии. В развитии ТМА при ВИЧ-инфекции не исключается роль неопластических процессов, возбудителей оппортунистических заболеваний, при которых повреждение эндотелия и/или агрегацию тромбоцитов могут вызывать многочисленные причины. Например, было показано, что цитомегаловирус (ЦМВ) увеличивает прокоагулянтную активность эндотелиальных клеток [15].
ВИЧ-ассоциированная ТМА чаще возникает у пациентов с низким уровнем CD4 лимфоцитов и высокими уровнями вирусной РНК. Лечение заключается только в поддерживающей терапии и назначении антиретровирусных препаратов.
ТМА, ассоциированная с аутоиммунными заболеваниями
ТМА может возникнуть у пациентов с аутоиммунными заболеваниями, такими как CКВ, КАФС, системная склеродермия.
Yu F. еt al., сравнив 55 случаев волчаночного нефрита классов IV-G и 7 случаев СКВ с ТМА, показали, что пациенты с СКВ и ТМА имеют более тяжелое почечное повреждение (по данным гистопатологического заключения) и, соответственно, худший почечный прогноз [16].
По данным N. Pattanashetti, в индийской популяции наличие ТМА также ассоциировалось с более тяжелой клинической картиной (высокие уровни протеинурии, более значимое повышение азотистых показателей) и худшим прогнозом [17].
Не существует стандартизированных протоколов лечения ТМА, ассоциированной с СКВ. Часто терапия включает глюкокортикоиды, иммуносупрессивную терапию, антикоагулянты, плазмаферез. К сожалению, данная терапия оказывается неэффективной более чем в 50% случаев.
В литературе описываются случаи успешного лечения экулизумабом [18].
Случаи КАФС, по международным данным, сопровождались ТМА в 14% случаев (всего было зарегистрировано 522 эпизода) [19]. Наиболее часто используемая терапия включала антикоагулянты, глюкокортикоиды, плазмообмен, внутривенное введение иммуноглобулина. Только 0,2% пациентов получали экулизумаб [19].
В нескольких исследованиях продемонстрировано успешное использование экулизумаба в качестве патогенетической терапии, т.к. в экспериментальных моделях на мышах показано вовлечение системы комплемента в патофизиологию КАФС [20].
ТМА, ассоциированная с гломерулярными заболеваниями
ТМА может быть ассоциирована с IgA-нефропатией (ИГАН – иммуноглобулиновая А нефропатия), мембранозной нефропатией, фокальным носегментарным гломерулосклерозом (ФСГС), АНЦА-ассоциированными васкулитами и МПГН/С3. Однако эта ассоциация может быть лишь патоморфологической находкой без клинико-лабораторной манифестации [20]. ИГАН при ассоциации с ТМА имеет худший прогноз. У таких пациентов более высокие уровни артериального давления и значимо выше протеинурия [21].
ТМА при АНЦА-ассоциированном васкулите не редкость и сопряжена с более тяжелым почечным повреждением (высокий уровень креатинина, клеточные полулуния, более тяжелое интерстициальное воспаление) [22].
Коллапсирующий вариант ФСГС является наиболее частой находкой при биопсии почек с ТМА. Следовательно, эндотелиальное повреждение играет важную роль в патофизиологии ФСГС. Наличие ФСГС ассоциировано с худшим почечным прогнозом без прогностического различия между коллапсирующим или иными вариантами ФСГС [23].
Описано несколько случаев развития коллапсирующего варианта ФСГС у пациенток с преэклампсией. Вероятным триггером являлась ишемия, развившаяся в результате ТМА. Плацента при преэклампсии увеличивает секрецию растворимой fms-подобной тирозинкиназы-1 (sFlt-1), ингибирущая рецепторы VEGF, что вызывает эндотелиальную дисфункцию и ТМА [24].
Эффективность экулизумаба при ТМА, ассоциированных с гломерулярными заболеваниями, не подтверждена [25].
ТМА после трансплантации сóлидных органов
ТМА были зафиксированы после трансплантации почек, печени, поджелудочной железы, легкого и сердца. Частота случаев составляет 2–4% [26]. При этом, по одним данным, ТМА возникает в первые 100 дней после трансплантации, по другим – в первые 3 месяца. Патогенез, вероятнее всего, имеет мультифакториальную природу: антителоопосредованное отторжение, ишемическое повреждение, вирусные инфекции (ЦМВ), иммуносупрессивные препараты (преимущественно ингибиторы кальциневрина).
В исследовании N. Kishida AB0-несовместимость при родственной трансплантации печени является независимым фактором риска развития ТМА (ТМА были зафиксированы в 37% случаев) [27]. Высказывались предположения, будто это связано с применением ритуксимаба, однако они не подтвердились.
Показатели выживаемости составляют около 70% после трансплантации печени, легких или кишечника и >80% – после трансплантации почки.
Повышение отношения фактор фон Виллебранда/ADAMTS13 имеет диагностическую значимость для пациентов, перенесших трансплантацию печени, однако показатель не валидирован, референсные значения точно не определены [28].
Острые состояния, такие как инфекция или реакция острого отторжения, повышают риск развития ТМА и ухудшают прогноз. Другими важными факторами риска развития ТМА являются комбинация ингибиторов кальциневрина с ингибиторами пролиферативного сигнала (mTOR), указание на перенесенную ТМА в прошлом, наличие антифосфолипидных аутоантител.
Лечение, направленное на ликвидацию повреждающего фактора (снижение дозы или отмена препарата, конверсия иммуносупрессивных препаратов, преимущественно ингибиторов кальциневрина и ингибиторов пролиферативного сигнала, лечение реакции отторжения и вирусных инфекций), может дать положительный результат.
Плазмаферез, использование свежезамороженной плазмы являются стандартом лечения ТМА в раннем периоде после трансплантации печени, но в случаях трансплантации других сóлидных органов использование плазмафереза остается спорным [28].
ТМА после трансплантации красного костного мозга
ТМА осложняет течение аллогенной трансплантации костного мозга в 10–40% случаев и сопряжено с высокой смертностью – до 21–75% случаев [11]. Медиана развития ТМА составляет 44 дня после трансплантации. К факторам риска относятся женский пол, тяжелое течение основного заболевания, инфекции, несовместимость c донором по системе HLA [28]. Почки поражаются редко. Природа ТМА в данном случае остается мультифакториальной, связанной с применением ингибиторов кальциневрина, повреждением аллографта, химиотерапией, лучевой терапией и инфекциями: ЦМВ, парвовирус, аденовирус, вирус гриппа А [29–32].
Лечение подобных состояний остается предметом изучения. Наиболее часто применяется плазмаферез, однако рациональность данного метода представляется спорной ввиду отсутствия циркулирующих ингибиторов факторов комплемента. В нескольких исследованиях показана эффективность экулизумаба [33]. Более того, исследование мутаций системы комплемента может позволить выявить пациентов высокого риска развития ТМА, но неясно, каким образом профилактировать данное состояние.
Важно отметить, что ТМА может поражать любую систему организма (без клинической манифестации). J. Schwimmer et al. проанализировали 742 почечные и почечно-поджелудочные трансплантации, выполненные в течение 15 лет, и выявили 21 гистологически подтвержденный случай ТМА [34]. В целом у 62% этих пациентов была констатирована симптоматика системной ТМА с МАГА и тромбоцитопенией, в то время как в 38% случаев ТМА была локализована только в графте.
Не последнюю роль должно играть генетическое тестирование мутаций системы комплемента.
Лекарственно-ассоциированная ТМА
В основе возникновения лекарственно-индуцированной ТМА лежат два механизма: иммунно-опосредованное повреждение и прямое токсичное (дозозависимое) действие (табл. 4) [35].

Иммунный механизм был впервые выявлен у пациента с рецидивирующими эпизодами ОПП, гемолиза и тромбоцитопении, вызванной приемом хинина. Воздействие препарата приводит к продукции антител, способствующих микроваскулярному повреждению и тромбоцитопении потребления. Механизм прямого токсического эффекта еще не полностью изучен, но, возможно, ключевым фактором является сниженная экспрессия сосудистого эндотелиального фактора роста и прямое эндотелиальное повреждение [36].
В тактике ведения основным этапом служит прекращение применения потенциально вредного препарата. Положительная динамика лабораторных показателей наблюдается в течение нескольких дней, в то время как почечная функция восстанавливается значительно медленнее, а в некоторых случаях почечный прогноз остается неудовлетворительным.
Метилмалоновая ацидурия с гомоцистеинурией
В основе данного заболевания лежит нарушение метаболизма кобаламина [37]. Необходимо исключать у детей в возрасте до 6 месяцев. Развитие данной патологии ассоциировано с мутациями в генах MUT, MMAA, MMAB, MMACHC, MMADHC, MCEE. Клинические проявления включают рвоту, развитие дегидратации, отказ от еды, снижение массы тела, генерализованную мышечную слабость, вялость, сонливость, коматозные состояния и судороги. При лабораторном исследовании выявляют тяжелый метаболический ацидоз, гиперглицинемию, гипераммониемию, гиперурикемию, повышение в сыворотке крови уровня пропионилкарнитина и снижение содержания свободного карнитина, повышение в моче концентраций метилмалоновой кислоты, 3-гидроксипропионовой, 3-гидрокси-n-валериановой, метилмалоновой кислот, гомоцистеина. Тактика терапии и коррекции сопутствующей ТМА основана на применении специализированных продуктов на основе аминокислот без изолейцина, валина, треонина и метионина, кофакторная терапия витамином B12, назначение левокарнитина и глицина [37].
ТМА, ассоциированная с беременностью
Преэклампсия и HELLP-синдром (Н – гемолиз, EL – повышенный уровень ферментов печени, LP – тромбоцитопения) являются специфическими, ассоциированными с беременностью формами ТМА. В структуре ТМА их доля составляет 8–18% от всех случаев [38–40]. ТМА, ассоциированная с беременностью, сопряжена с высокой смертностью (до 10%) [41].
Всем пациенткам, госпитализированным с диагнозом «тяжелая преэклампсия» и/или «HELLP-синдром», необходимо еще до родоразрешения исследовать лактатдегидрогеназу, гаптоглобин в сыворотке крови и шизоциты в мазке периферической крови, а также определить число тромбоцитов и уровень креатинина. При подозрении на акушерскую ТМА следует провести дифференциальную диагностику с аГУС, ТТП, КАФС, острой жировой печенью беременных. аГУС обычно возникает после родов (80%), ТТП – и в до-, и в послеродовом периодах.
Акушерская ТМА – одна из важнейших причин развития синдрома полиорганной недостаточности при беременности и после родов [42].
Дифференциальный диагноз
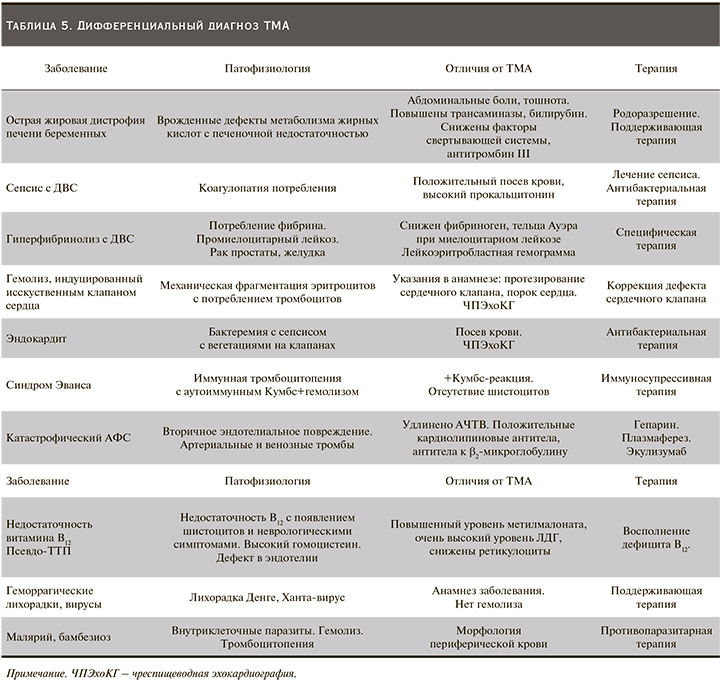
Дифференциальную диагностику ТМА следует проводить с целым спектром заболеваний.
В табл. 5 обобщены сведения о патогенезе, лечении и отличительных от ТМА признаках различных состояний.
Заключение
ТМА – клинико-морфологический синдром, относящийся к целому спектру заболеваний с эндотелиальным повреждением в качестве патогенетического фактора.
Клиническая картина вторичных ТМА не имеет специфических черт. Условием постановки клинического диагноза ТМА является наличие как минимум двух симптомов: тромбоцитопении и МАГА.
Развитие вторичных ТМА связано с широким спектром различных заболеваний и состояний, рассмотренных в данном обзоре. Наиболее важно вовремя распознать их, т.к. это значительно улучшает прогноз. Своевременная диагностика и лечение ТМА, ассоциированной с беременностью, показывют хорошие результаты. С другой стороны, плохой прогноз при ТМА, ассоциированной с трансплантацией гемопоэтических стволовых клеток, и ТМА, ассоциированной с химиотерапией.


