
Эпидемиология
Первое описание массивных отложений IgM в клубочках у больных гематурическими формами нефрита было сделано L.B.A. Van de Putte и соавт. в 1974 г. [1]. Позже A.H. Cohen и соавт. (1978) и H.K. Bhasin и соавт. (1978) описали это заболевание у больных протеинурической формой нефрита [2, 3]. Спустя несколько лет случаи IgM-нефропатии были описаны в Англии, Канаде, Японии, а также на Тайване [4–7].
Распространенность IgM-нефропатии, по данным нефрологических регистров, колеблется от 2 до 5 % от общего числа всех случаев гломерулонефрита [8–10]; в общей популяции она составляет около 0,3 случая на 1 млн населения [8]. Отдельные исследования демонстрируют очень высокую (45,8 %) частоту IgM-нефропатии [11], однако при этом нельзя исключить гипердиагностику, связанную с не вполне адекватной трактовкой результатов иммуногистохического анализа почечных биоптатов.
В Российской Федерации исследования по проблеме IgM-нефропатии немногочисленны, до сих пор отсутствуют детальные клинические и морфологические описания случаев болезни. Имеются данные о распространенности IgM-нефропатии, полученные в ходе клинико-морфологических исследований когорт больных хроническим гломерулонефритом. Так, по данным П.Н. Любченко и соавт., распространенность IgM-нефропатии составляет 3,8 % [12]. В исследовании Н.Н. Коряковой в 54,2 % случаев мезангиопролиферативного гломерулонефрита регистрировалась IgM-нефропатия [13].
В ряде нефрологических регистров производилась оценка распространенности тех случаев нефритов, при которых регистрируется диффузное свечение IgM или IgG без разделения по типам свечения. В частности, в описании данных сербского регистра приведены данные о распространенности неIgA-нефропатии (мезангиопролиферативного гломерулонефрита), которая составила 8,7 % [14]. В эту группу вошли пациенты и с IgM-нефропатией. Исследователями приведены также данные о распространенности неIgA-нефропатии начиная с 1987 г., что позволило оценить динамику процесса. Пик распространенности неIgA-нефропатий приходился на 1992–1997 гг., когда значения данного показателя достигали 25–34 %. При анализе данных испанского регистра с 1994 по 1999 г. существенных колебаний в распространенности неIgA-нефропатий не было установлено, колебания составили 3,3–6,6 % [15]. Было показано, что удельный вес неIgA-нефропатий выше в детском возрасте (9 %), далее снижается у взрослых (5,8 %) и несколько повышается в старшей возрастной группе (6,1 %). Вместе с тем, по данным исследования L.F. Arias и соавт., удельный вес IgM-нефропатий в детской когорте больных гломерулонефритами был несколько ниже и составил 5,17 % [16].
По данным чешского регистра, удельный вес неIgA-нефропатий составляет 11,3 % [17]. Гендерных различий в ходе анализа распространенности IgM-нефропатии выявлено не было [10, 16].
Большинство описаний касается возникновения IgM-нефропатии в нативной почке, описаны единичные случаи развития нефрита в почечном трансплантате [18].
Этиология и патогенез
Этиология IgM-нефропатии не известна. Тем не менее частое выявление комплементарных депозитов (C4d, С3) в почечных клубочках при IgM-нефропатии, вероятно, свидетельствует о том, что IgM является компонентом классического иммунного комплекса, медитирующего активацию комплементарного каскада в ответ на повреждение [10, 16, 19, 21]. Вместе с тем не известна антигенная природа иммунной реакции, лежащей в основе заболевания [22]. Предположительно антигенами могут выступать субстанции экзогенного происхождения, в т.ч. пищевые [5, 9, 12, 23].
Как известно, IgM имеют низкую аффинность к специфическим микробным патогенам, как правило, полиреактивны и отвечают в большей степени за инициацию распознавания инфекционных агентов, включая бактерии, вирусы и модифицированные аутоантигены [25]; в последующем ускоряется генерация высокоаффинных IgG. Низкая аффинность IgM обусловлена наличием множественных Fab-участков [26]. IgM экспрессируются в качестве трансмембранных мономерных форм на всех нативных В-клетках. Известно три формы секреторных IgM – монометрическая, пента- и гексаметрическая. Высокий уровень именно монометрических форм обнаруживается при аутоиммунных заболеваниях, таких как системная красная волчанка, ревматоидный артрит, смешанная криоглобулинемия, некоторые формы гломерулонефритов, в т.ч. при IgM-нефропатии, а также злокачественных лимфопролиферативных синдромах [27].
При большинстве форм гломерулонефрита, в т.ч. при IgM-нефропатии, плазменные уровни IgM в большинстве случаев не изменены, концентрация IgM в депозиты происходит на тканевом уровне. Предполагают, что данную патологию можно будет отнести к известной группе гипер-IgM-синдромов, характеризующихся генетическим дефектом CD40L–CD40-путей [28]; однако с клинической точки зрения эта гипотеза пока не подтверждена – имеются лишь единичные описания случаев семейной IgM-нефропатии [29].
По мнению A. Vanikar, в патогенезе IgM-нефропатии определенную роль играет дисфункция Т-клеточного звена, проявляющаяся гиперфункцией Т-супрессоров [22]. В целом IgM-нефропатию считают идиопатическим первичным гломерулярным заболеванием.
Патоморфология
Единых критериев постановки диагноза по биопсиям до сих пор нет, что усложняет оценку распространенности, клинико-морфологических особенностей и прогноза этого заболевания. Основой диагноза служит выявление диффузных глобальных мезангиальных депозитов IgM при иммунофлюоресценции, интенсивность сигнала выше умеренной ((≥ 2+). В 30–100 % случаев возможно также выявление депозитов С3. Отдельные авторы описывают слабое неспецифическое свечение до 1+ IgG, IgA, и C1q [10]. Гистологическая картина IgM-нефропатии может быть полиморфной. При световой микроскопии клубочки могут выглядеть нормально, иногда наблюдаются умеренная сегментарная мезангиальная гиперклеточность, умеренный мезангиальный склероз, фокальный сегментарный гломерулосклероз (ФСГС) [10]. При электронной микроскопии отмечаются диффузное исчезновение малых отростков подоцитов и аморфные мезангиальные слабо электронно-плотные депозиты. Эти изменения позволяли ряду исследователей трактовать IgM-нефропатию как вариант болезни минимальных изменений [30, 31]. Дискуссионным остается вопрос и о связи ФСГС и IgM-нефропатии. Часть исследователей отталкиваются от основного критерия поставки диагноза – глобального выявления в клубочке IgM депозитов при флюоресценции, а сегментарное их распределение предлагают считать признаком идиопатической формы ФСГС. Другие авторы указывают, что определяющим является интенсивность реакции, а не распределение депозитов, поэтому ФСГС с интенсивностью сигнала выше умеренной ((≥ 2+) должен трактоваться как IgM-нефропатия [31, 32].
Мы располагаем тремя наблюдениями IgM-нефропатии. Пациенты – двое мужчин и одна женщина в возрасте 20 и 23 года, 31 года соответственно. У всех пациентов в течение нескольких месяцев наблюдалось развитие нефротического синдрома (протеинурия свыше 3–4 г/л, отеки вплоть до анасарки), гематурии, у одного больного – умеренное повышение артериального давления.
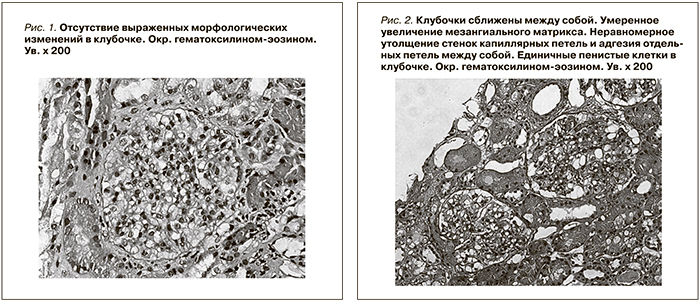
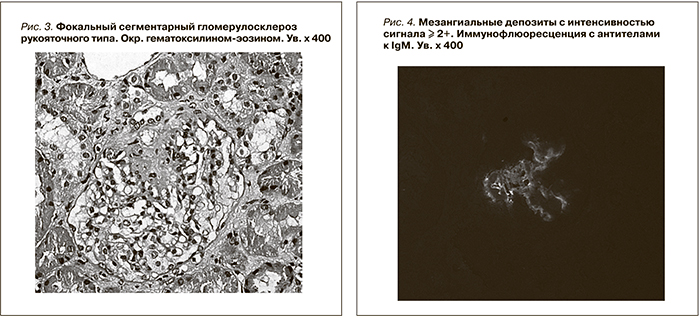
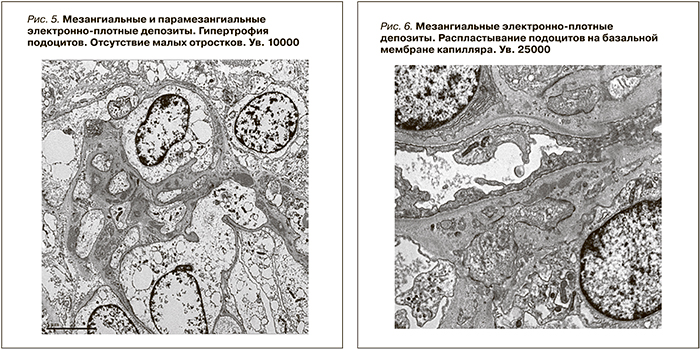
При световой микроскопии большинство клубочков в биоптатах выглядело нормальным (рис. 1). Только в отдельных клубочках присутствовали начальные признаки ФСГС в виде неравномерного утолщения стенок отдельных капиллярных петель, их сращение между собой (рис. 2), либо признаки ФСГС по рукояточному типу (рис. 3). Во всех наблюдениях имелись признаки хронического тубулоинтерстициального нефрита в виде очаговой слабой или умеренно выраженной лимфо-гистио-плазмоцитарной инфильтрации и фиброза интерстиция. При иммунофлюоресценции выявлены глобально или сегментарно расположенные мезангиальные и парамезангиальные депозиты IgM, дающие интенсивность сигнала выше умеренной ((≥ 2+) (рис. 4). В двух наблюдениях отмечена коэкспрессия С3-позитивных депозитов с интенсивностью сигнала выше 2+. При электронной микроскопии определены немногочисленные слабой электронной плотности мезангиальные и парамезангиальные депозиты. Подоциты на всем протяжении капиллярных петель были гипертрофированы, лишены малых отростков, распластаны на базальной мембране. В цитоплазме содержались вакуоли липидов, отмечалась микровиллезная трансформация клеток (рис. 5, 6).
Кроме того, мы наблюдали сочетанное отложение депозитов IgA и IgM у пациента в возрасте 21 года. В течение 4 месяцев у него отмечены признаки нарастающего нефротического синдрома с суточной протеинурией до 4 г/сут, анасаркой. Пациент страдал также микрогематурией и умеренным повышением артериального давления. При световой микроскопии биоптата выявлено умеренное расширение мезангиального матрикса и слабовыраженная сегментарная мезангиальная гиперклеточность. При иммунофлюоресценции определены глобально расположенные мезангиальные депозиты IgA и IgM, дающие интенсивность сигнала выше умеренной ((≥ 2+). При электронной микроскопии обнаружены мезангиальные и парамезангиальные электронно-плотные депозиты, отсутствие малых отростков подоцитов с распластыванием клеток на базальной мембране. Данное наблюдение сходно с описанными тремя наблюдениями J.T. Mustonen и соавт., предполагающими, что это может быть особой подгруппой первичных гломерулопатий – IgA-IgM-нефропатией [33].
Клиническая картина
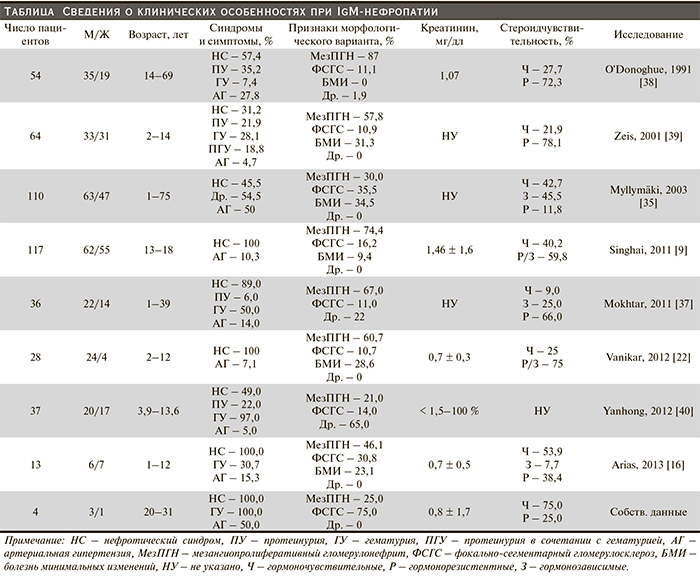
Нефротический синдром является наиболее частым клиническим проявлением IgM-нефропатии в группе как детей [34], так и взрослых [10, 35]. Риск развития терминальной почечной недостаточности колеблется от 4 до 23 % [35–37]. Артериальная гипертензия формируется в 24,4–50,0 % клинических наблюдений [10, 35]. Гематурия сопутствует нефротической протеинурии в половине наблюдений [10]. Сведения о клинических особенностях при IgM-нефропатии представлены в таблице.
По данным исследователей течение нефрита отличается меньшими темпами прогрессирования почечной дисфункции в случае латентных форм, проявляющихся гематурией. Протеинурия и артериальная гипертензия служат фактором менее благоприятного прогноза. Обнаружение при нефробиопсии признаков ФСГС обычно свидетельствует о высоком риске быстрого прогрессирования нефрита с формированием терминальной почечной недостаточности [9, 22, 35]. В большинстве исследований показано, что появление депозитов IgM при болезни минимальных изменений также является признаком более тяжелого прогноза как в детской, так и во взрослой популяции больных [9, 22, 39]. Однако в исследовании A.F. Donia и соавт. IgM-свечение негативно влияло на почечную функцию при болезни минимальных изменений у детей только в случае сочетания его с мезангиальной гиперклеточностью [41]. Таким образом, существующая клиническая и морфологическая неоднородность IgM-нефропатии определяет различия в темпах прогрессирования потери почечной функции.
Лечение
Глюкокортикоиды остаются основным препаратом лечения IgM-нефропатии в случае формирования нефротического синдрома. Примерно в трети случаев нефротический синдром стероидрезистентен (см. таблицу). Стероидзависимость при IgM-нефропатии развивается приблизительно в 80 % случаев [35]. Методы терапии при течении IgM-нефропатии с нефритическим синдромом не разработаны, и обычно применяются технологии, эффективность которых описана при ФСГС, мезангиопролиферативном нефрите, т.е. тех формах, морфологические признаки которых регистрируются при IgM-нефропатии. В отдельных исследованиях фигурируют данные об эффективности цитостатиков, применяемых при нефротических формах совместно с глюкокортикоидами. Однако эти данные малочисленны для того, чтобы стать поводом углубленного анализа и рекомендаций по терапии при IgM-нефропатии. В частности, в исследовании L.F. Arias и соавт. 3 (23,1 %) детей с IgM-нефропатией получали циклофосфамид, 5 (38,4 %) – мофетила микофенолат, 1 (7,7 %) ребенок – циклоспорин [16]. Основные причины применения цитостатиков: частые рецидивы нефротического синдрома или стероидрезистентность. В 5 из 9 случаев добиться ремиссии нефротического синдрома в течение года терапии не удалось. В исследовании K. Kanemoto и соавт. 14 детей со стероидрезистентным нефритом получали циклоспорин и всеми больными была достигнута парциальная или полная ремиссия IgM-нефропатии [42]. Описаны единичные случаи применения ритуксимаба при IgM-нефропатии с положительным эффектом [43].
Резюмируя вышеизложенное, следует заметить, что лечение IgM-нефропатии производится исходя из клинической картины нефрита. При наличии нефротического синдрома основным способом лечения служит применение глюкокортикоидов, в случае их неэффективности или частых обострений рекомендуется комбинация их с цитостатиками (циклофосфамид, циклоспорин). При выборе цитостатика, вероятно, следует ориентироваться на морфологические проявления и в случае регистрации признаков ФСГС или болезни минимальных изменений выбор делается в пользу циклоспорина, в случае иных морфологических проявлений – циклофосфамида. Вариант течения с нефритическим или изолированным мочевым синдромом предполагает негормональную терапию с применением блокаторов ренин-ангиотензин-альдостероновой системы. Данный класс препаратов рекомендуется применять также в сочетании с гормональной терапией при нефротическом варианте нефрита.
Таким образом, выделение IgM-нефропатии в качестве варианта первичного гломерулонефрита объясняется особенностями клинической картины болезни и прогрессирования почечной дисфункции при диффузном отложении депозитов IgM. Вместе с тем низкая распространенность данной патологии не позволяет в настоящее время с уверенностью говорить о медикаментозных возможностях хорошего контроля за течением заболевания. Изложенные сведения о четырех случаях IgM-нефропатии, один из которых – IgA-IgM-нефропатия, позволяют расширить представления о клинико-морфологической картине IgM-нефропатии.


