
Введение
Почкам принадлежит важная роль в поддержании баланса кальция и фосфора. При почечной недостаточности нарушаются все звенья фосфорно-кальциевого обмена. Уже при снижении скорости клубочковой фильтрации (СКФ) ниже 60 мл/мин/1,73 м2 уменьшается фильтрация фосфора и
повышается его сывороточная концентрация, что вызывает повышение секреции ПТГ. ПТГ подавляет реабсорбцию фосфора, таким образом нормализуя его уровень в сыворотке крови, но при падении СКФ ниже 30 мл/мин/1,73 м2 этот механизм поддержания нормальной сывороточной концентрации фосфора становится недостаточно эффективным и развивается стойкая гиперфосфатемия, что стимулирует усиленную секрецию ПТГ. При гиперфосфатемии ингибируется активность 1α-гидроксилазы в проксимальных канальцах, снижются продукция и содержание в сыворотке
крови 1,25(OH)2D3 — кальцитриола. Дефицит кальцитриола вызывает нарушение всасывания кальция в тонком кишечнике и развитие гипокальциемии. При гипокальциемии, персисстирующей в течение месяцев, развивается гиперплазия паращитовидных желез (ПЩЖ), обусловливающая избыточную
продукцию и секрецию ПТГ, что наряду с гиперфосфатемией является проявлением вторичного гиперпаратиреоза (ВГПТ). Гипокальциемия, дефицит витамина D и гиперфосфатемия – самые важные факторы, ответственные за гиперплазию ПЩЖ. Гипокальциемия, относительный или абсолютный дефицит кальцитриола, гиперплазия ПЩЖ могут развиваться на начальном этапе нарушения функции почек – хроническая болезнь почек (ХБП) 3-й стадии (СКФ – 60–30 мл/мин/1,73 м2) и прогрессировать по мере нарастания почечной недостаточности – ХБП 4–5-й стадий (СКФ – 29–15 мл/мин/1,73 м2).
Нарушения фосфорно-кальциевого обмена при ХБП вызывают патологию костной системы, которую обозначают термином “ренальная остеодистрофия” (РОД).
Патогенез нарушений фосфорно-кальциевого обмена у больных ХБП III–V стадий
Гиперфосфатемия. Уровень сывороточного фосфора определяется соотношением между абсорбцией фосфора из желудочно-кишечного тракта (ЖКТ), мобилизацией его из костей (резервуар фосфора и кальция) и выведением почками. С едой человек потребляет примерно 1000–1200 мг фосфора, 800 мг из этого количества абсорбируется в кишечнике, составляя т. н. обменный пул фосфора. Обменный пул фосфора в организме содержится в цитоплазме клеток (70 %), в костях (29 %) и сыворотке крови (менее 1 %). Баланс фосфора в организме регулируется в основном почками. В норме почки фильтруют около 9 г фосфора, из которых около 8 г. (90 %) реабсорбируются в проксимальных канальцах с помощью трех транспортеров неорганического фосфора: Na+/ Pi-котранспортеры I, II и III типов [1, 2]. Транспортеры I и II типов локализованы на апикальной мембране канальцевого эпителия, а III – на базальной мембране. Реабсорбция фосфора сопряжена с транспортом Na+ и зависит от количества
молекул Na+/Pi-котранспортера на апикальной мембране эпителия проксимальных канальцев [2].
Высокое содержание фосфора во внеклеточной жидкости подавляет 1αOHD3-гидроксилазу — фермент, превращающий 25(OH)D3 в 1,25(OH)2D3, а низкое содержание фосфора приводит к противоположному эффекту. Влияние внеклеточного фосфора на активность фермента не зависит от
ПТГ и Na+/Pi-котранспортера. При ХБП может развиваться и гипофосфатемия, например, вследствие синдрома нарушенного всасывания, злоупотребления фосфат-биндерами, гипервентиляции, авитаминоза D, длительного применения глюкокортикоидов. Хронический дефицит фосфата повышает его реабсорбцию в почках за счет образования новых молекул Na+/Pi-котранспортера. Одновременно в почках увеличивается продукция 1,25(OH)2D3 и повышается концентрация Са2+ в крови. За счет увеличения всасывания Са2+ в кишечнике и реабсорбции в почках подавляется секреция ПТГ и увеличивается почечная реабсорбция фосфора. Это свидетельствует о тесном взаимодействии факторов, регулирующих транспорт фосфора и кальция. Кальцитриол увеличивает абсорбцию фосфора за счет интенсификации его захвата пузырьками щеточной каемки энтероцитов [1–3]. Персистирующая гиперфосфатемия может выявляться уже при падении СКФ ниже 60 мл/мин/1,75 м2 . При более выраженном снижении функции почек (СКФ ниже 30 мл/мин/1,73 м2) развивается постоянная гиперфосфатемия. Ретенция фосфора при ХБП может способствовать гипокальциемии не только посредством прямого влияния гиперфосфатемии на сывороточный уровень кальция, но и на функцию паращитовидных желез. Экспериментально доказано, что гиперфосфатемия может стимулировать гиперплазию ПЩЖ независимо от гипокальциемии и/или дефицита витамина D3. Поэтому у больных ХБП содержание фосфора в сыворотке крови следует поддерживать на уровне 2,7–4,6 мг/дл (0,87–1,49 ммоль/л), у больных ХБП 5-й стадии – в диапазоне 3,5–5,5 мг/дл (1,13–1,78 моль/л) [3–5].
Гипокальциемия. В норме взрослые ежедневно потребляют с пищей 1,0–1,5 г кальция. Большая часть его всасывается в двенадцатиперстной кишке и проксимальных отделах тощей кишки. Абсорбция кальция зависит от его содержания в пище и степени минерализации скелета. Кальций лучше всасывается при приеме его препаратов во время еды [1, 2, 5]. Около 5–7 г/сут кальция фильтруется в клубочках почек, из которых 95–97,5 % реабсорбируются в канальцах [1]. Основным депо кальция
является костная ткань, в которой сконцентрировано до 98 % кальция организма. В сыворотке крови половина общего содержания кальция находится в свободном (ионизированном) виде, треть – в комплексе с белками плазмы, в основном с альбумином, а остальная часть – в комплексе с другими катионами. От содержания в крови ионизированного кальция (Ca2+) зависят все биологические эффекты кальция. При развитии ХБП 4–5-й стадий нарушение всасывания кальция в ЖКТ приводит к
снижению в крови общего и ионизированного кальция [2, 5]. Основные источники кальция – молочные и мясные продукты. Поступающий с пищей кальций связан с белком, в ЖКТ под действием протеаз кальций высвобождается из-под связи с белком. Скорость абсорбции Ca2+ в двенадцатиперстной кишке определяет концентрация кальцийсвязывающего белка – кальбиндина в цитоплазме энтероцитов двенадцатиперстной кишки. Активный транспорт Ca2+ регулируется кальцитриолом,
оказывающим биологический эффект через взаимодействие со специфическим внутриклеточным рецептором, регулирующим транскрипцию более 60 генов, кодирующих кальбиндин и белки Ca2+-насоса [6–7]. Факторы, влияющие на абсорбцию кальция в кишечнике, представлены в табл. 1.
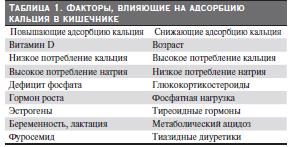
У здорового человека суточная экскреция Ca2+ составляет 40–300 мг [1, 2]. Гиперфосфатемия приводит к снижению плазменного уровня ионизированного кальция за счет как прямого связывания, так и снижения продукции почками 1,25(OH)2D3. Кальций – самый важный регулятор секреции
ПТГ. Даже кратковременная гипокальциемия вызывает увеличение секреции ПТГ за счет активации Са-рецепторов, расположенных на ПЩЖ. При персистирующей гипокальциемии увеличиваются уровень матричной РНК ПТГ и транскрипция гена пре-про-ПТГ, сопровождающаяся увеличением продукции ПТГ, а в последующем – гиперплазией ПЩЖ, обеспечивающей усиленную продукцию и высвобождение ПТГ. При гипокальциемии ПТГ стимулирует продукцию 1,25(OH)2D3 в ткани почек, усиливающего абсорбцию кальция из ЖКТ [4, 5]. При ХБП IV–V стадий обратная зависимость между
уровнем кальция и секрецией ПТГ нарушается, в результате требуется более высокое, чем у здоровых лиц, содержание ионизированного кальция для угнетения продукции ПТГ. Поэтому желательно поддерживать сывороточный уровень ионизированного кальция в пределах верхней границы нормы
[1, 2]. Концентрация кальция в диализате у больных на ГД и ПД должна быть 2,5 мэкв/л (1,25 ммоль/л). У больных ХБП 4–5-й стадий уменьшается чувствительность аденилатциклазы паращитовидных желез к ингибирующему влиянию кальция. Угнетают абсорбцию кальция ГКС, ингибируя превращение
25OHD3 в 1,25(OH)2D3, а также фосфаты, образующие с ним нерастворимые комплексы, и оксалаты [1].
Гиперпродукция ПТГ и кальцитонина. Молекула ПТГ состоит из 84 аминокислотных остатков (1–84), включает укороченный N-концевой фрагмент (ПТГ 1–34) и длинный С-концевой фрагмент (ПТГ 7–84). Молекулярная масса ПТГ – 9425 Да. Ген, ответственный за синтез ПТГ, локализован на 11-й хромосоме. Биологической активностью обладает не только вся молекула ПТГ, но и его N-концевой (терминальный) фрагмент [11–12].
ПТГ отвечает за регуляцию уровня Ca2+ в крови. Под действием этого гормона концентрация Ca2+ в крови увеличивается. В свою очередь при повышении концентрации кальция к крови синтез ПТГ снижается [11–14]. Благодаря такой обратной связи концентрация Ca2+ в крови поддерживается
на постоянном уровне.
ПТГ участвует в поддержании постоянства концентрации Ca2+ в крови:
• стимулируя резорбцию костной ткани, вызывая выход кальция в кровь;
• усиливая реабсорбцию кальция в почках, снижая его выведение с мочой;
• увеличивая всасывание кальция в тонкой кишке (опосредованно – путем стимуляции синтеза 1,25(ОН)2D3).
Изменения концентрации кальция контролируются в первую очередь за счет влияния ПТГ на костную ткань и в меньшей степени – на выведение кальция почками. Долговременное поддержание кальциевого баланса осуществляется в основном за счет действия ПТГ на синтез 1,25(ОН)2D3 и, следовательно, на всасывание кальция в ЖКТ. Ежедневно между кровью и костной тканью обменивается до 500 мг кальция. ПТГ – главный регулятор этого обмена [15].
ПТГ оказывает ряд прямых и опосредованных эффектов на костную ткань. Постоянное повышение уровня ПТГ приводит к увеличению числа клеток костной ткани, особенно остеокластов, и к интенсификации процессов перестройки (ремоделирования) кости. Скорость секреции ПТГ в ПЩЖ
зависит в основном от уровня Ca2+ в крови. Магний обладает сходным с кальцием, но менее выраженным действием на секрецию ПТГ. Физиологические колебания магния практически не влияют на секрецию ПТГ, но при выраженном снижении внутриклеточного содержания магния секреция
ПТГ повышается [15, 16].
Ca2+ влияет на секрецию ПТГ через взаимодействие с Сa-рецепторами (CaR), сопряженными с G-белками и имеющими большой внеклеточный домен для связывания низкомолекулярных лигандов. Активация рецепторов при высоком уровне Ca2+ в крови подавляет секрецию ПТГ через систему
вторых посредников – инозитол-1,4,5-трифосфат (ИФ3) и 1,2-диацилглицерин. CaR обнаружены в клетках ПЩЖ, в С-клетках щитовидной железы, секретирующих кальцитонин, в клетках головного мозга и почек. С-концевые фрагменты оказывают не резорбирующий, а анаболический эффект на
костную ткань [1, 2]. В норме ПТГ и его фрагменты выводятся из организма почками и метаболизируются в клетках проксимальных канальцев. ПЩЖ секретируют как целые молекулы
ПТГ, так и их фрагменты. Целые молекулы ПТГ быстрее элиминируются из крови, чем С-концевые фрагменты. При ХБП падение СКФ и нарушение метаболической функции почек приводят к накоплению в крови преимущественно С-концевых фрагментов. С помощью тестов I поколения для
определения ПТГ не различаются интактный ПТГ (иПТГ) и его фрагменты, результаты анализа отражают в основном концентрации неактивных фрагментов. В последнее время для измерения концентрации иПТГ используют иммунорадиометрический метод с двойным набором антител против
эпитопов N-терминального и С-концевого фрагментов, выявляя молекулу иПТГ. Плазменная концентрация и ПТГ повышается при ХБП 4–5 стадий, в первую очередь вследствие увеличения его секреции. Повышение секреции иПТГ обусловлено увеличением уровня Pi и снижением сывороточного уровня Ca2+ в сыворотке крови [2]. Основная функция иПТГ – поддержание постоянной концентрации Ca2+ во внеклеточной жидкости. иПТГ взаимодействует с рецептором, сопряженным с мембраносвязанной аденилатциклазой в проксимальных и дистальных канальцах почек, вызывает фосфатурию и гипофосфатемию, повышает реабсорбцию кальция в дистальных канальцах [1, 12–14]. Одновременно иПТГ стимулирует синтез 1,25(OH)2D3 в паренхиме почек. Снижение чувствительности
костной ткани к кальциемическому действию иПТГ отмечается уже при начальном снижении СКФ < 85 мл/мин/1,73 м2) и не регрессирует во время лечения ГД; его также выявляют и у многих реципиентов почечного трансплантата со сниженной почечной функцией (СКФ (ниже) < 70 мл/мин/1,73 м2),
а также у больных острой почечной недостаточностью (ОПН). У больных ОПН почти всегда наблюдается гипокальциемия. Гипокальциемия появляется на стадии олигурии и сохраняется
на всем протяжении периода полиурии. Гипокальциемия регрессирует после восстановления функции почек. Резистентность скелета к кальциемическому действию иПТГ связывают с дефицитом витамина D3, а также сниженной регуляторной активностью рецепторов иПТГ (PTH-PTHrP). ПТГ действует
на остеогенез через активацию остеобластов, а на резорбцию кости – остеокластов. Однако у остеокластов нет рецепторов для ПТГ, поэтому стимуляция остеокластов осуществляется опос-
редованно – через высвобождение остеобластами цитокинов. В эксперименте стимуляция резорбции кости остеокластами под действием ПТГ наблюдалась только в случае, если в культуре клеток имеются и остеокласты, и остеобласты [14–15]. Среди цитокинов ведущую роль отводят IGF-1, ИЛ-6, G-CSF, вызывающим пролиферацию и деференцировку предшественников остеокластов. Образуются крупные многоядерные остеокласты, которые начинают секретировать органические кислоты и гидролитические ферменты, растворяющие минеральную структуру и резорбирующие органическую основу костной ткани с высвобождением фосфора, кальция и бикарбоната во внеклеточную жидкость. Остеобласты синтезируют органические компоненты клеток (преимущественно коллаген I типа), но иПТГ процессы резорбции активирует в большей степени, чем синтеза кости. При развитии гипокальциемии ПТГ выводит кальций из костной ткани для поддержания постоянной концентрации кальция во внеклеточной жидкости. Даже небольшое повышение уровня иПТГ приводит к активации зрелых остеокластов и резорбции костной ткани [11, 12, 14, 15].
Кальцитонин секретируется в межфолликулярных клетках щитовидной железы. Его содержание в сыворотке крови повышено у 30 % больных терминальной стадией ХБП. Кальцитонин оказывает фосфатурический эффект, угнетая реабсорбцию фосфатов в проксимальных извитых канальцах,
а также повышает активность 1α-гидроксилазы, стимулируя продукцию 1,25(OH)2D3. Кальцитонин снижает концентрацию Ca2+ в сыворотке крови и внеклеточной жидкости, уменьшает количество и активность остеокластов в костной ткани, ингибируя остеолиз [1, 2].
Дефицит витамина D3. Кальцитриол (1,25-дигидроксихолекальциферол) образуется из витамина D3 (холекальциферола) путем его гидроксилирования (присоединения группы ОН) в положении 25 в печени и в положении 1 в проксимальных извитых канальца почек при участии α1-гидроксилазы. В почках кальцитриол индуцирует реабсорбцию Ca2+ и Pi. В тонком кишечнике под влиянием кальцитриола в ворсинках увеличивается продукция кальцийсвязывающего белка, повышающего абсорбцию Ca2+. В костной ткани кальцитриол стимулирует пролиферацию и дифференцировку преостеокластов. Под действием кальцитриола возрастает синтез компонентов органического матрикса и регулируются процессы минерализации костной ткани [16, 17]. У больных ХБП 3-й стадии может отмечаться относительный дефицит 1,25(OH)2D3. В дальнейшем развивается абсолютный дефицит кальцитриола, если уровень СКФ падает ниже 50 мл/мин/1,73 м2 у детей и ниже 30 мл/мин/1,73 м2 у взрослых [16–18]. Прогрессирование ХБП сопровождается уменьшением числа рецепторов витамина D (VDR) и Ca-рецепторов паращитовидных желез с падением чувствительности желез к действию 1,25(OH)2D3 и Ca2+. У пациентов с ХБП 4–5-й стадий плазменная концентрация 1,25(OH)2D3 снижена, что обусловлено нарушением его гидроксилирования в почках. У таких больных имеется и дефицит 1,25(OH)2D3, и устойчивость к его действию. У ренопривных больных и пациентов на диализе уровень 1,25(OH)2D3 в крови обычно не определяется современными диагностическими тестами. Уровень 25-гидроксивитамина D [25(OH)D3] в крови может быть низким у больных ХБП с протеинурией нефротического уровня из-за потерь 25(OH)D3 с мочой, у больных на перионеальном диализе – вследствие диффузии ПТГ в перитонеальный раствор и у пациентов с дефицитом витамина D в пище. Ограничение приема фосфора с пищей по мере снижения СКФ у пациентов с ХБП сопровождается увеличением в крови содержания 1,25(OH)2D3 и повышением ответа органов мишеней на воздействие кальцитриола. Кальцитриол подавляет активность ПЩЖ, действуя непосредственно на ген ПТГ; вызывает уменьшение его транскрипции и синтеза ПТГ; повышает чувствительность кальциевых рецепторов в клетках ПЩЖ. В экспериментальных работах показано, что дефицит кальцитриола может инициировать ВГПТ даже в отсутствие гипокальциемии. Дефицит кальцитриола ведет к нарушению абсорбции кальция в тонком кишечнике и снижению кальциемического действия на иПТГ. Гипокальциемия вызывает в свою очередь ВГПТ [1, 17–19].
Эктопическая минерализация при ХБП – механизмы и патогенез. Многие авторы рассматривают гиперфосфатемию при ХБП как особый синдром с уникальными особенностями костного ремоделирования, развитием гетеротопной минерализации и сердечно-сосудистых осложнений [1, 2, 18, 19]. Установлено, что среди больных ХБП с гиперфосфатемией частота АГ и средние уровни систолического и среднего давления достоверно выше, чем у больных с нормальным уровнем
фосфора в плазме. При этом компенсаторные механизмы сосудистой адаптации нарушены, поэтому высокое давление может приводить к большой механической нагрузке и потере эластичности артериальной стенки [1, 19]. Обнаружена прямая статистически значимая корреляция между выраженностью гиперфосфатемии и степенью ГЛЖ, а также между высоким произведением Са × Р (более 70 мг2/дл2) и частотой случаев гипертрофической кардиомиопатии (КМП) [19]. Последняя
наряду с ИБС является основной причиной хронической сердечной недостаточности у больных ХБП. Кроме того, высокая концентрация фосфора способствует неатеросклеротической кальцификации артерий вследствие трансформации гладкомышечных клеток сосудов (VSMC) в остеогенный фенотип [18–19].
В развитие гиперфосфатемии при ХБП вносит свой вклад ремоделирование костной ткани. При всем многообразии вариантов костного ремоделирования у пациентов с ХБП их объединяют общие изменения, характеризующиеся избыточной резорбцией кости по отношению к костеобразованию. В эксперименте установлено, что в условиях гиперфосфатемии функция скелета как резервуара фосфата блокируется [1, 19]. Однако при этом потребность костной ткани в фосфоре, напротив,
увеличивается, что стимулирует повышение его концентрации в крови, а новым местом для его депонирования становятся мягкие ткани и сосуды. Депозиты фосфора, обнаруживаемые в артериях, приводят к повышению жесткости (ригидности) сосудистой стенки [1, 18, 19].
Результаты исследований последних лет свидетельствуют, что больные ХБП с гиперфосфатемией (> 6,5 мг/дл) имели более высокий риск смерти в результате сердечно-сосудистых заболеваний, включая ИБС, чем те, у которых уровень фосфора был < 6,5 мг/дл. Принимая во внимание эти данные, контроль
уровня фосфата сыворотки < 6,5 мг/дл становится важной терапевтической задачей у таких пациентов [20–23].
При ХБП выявляют две формы сосудистой кальцификации: атеросклеротических неоинтимальных бляшек (интимальную) и медии артерий (медиальную) [20]. Обе формы сосудистой кальцификации прогрессируют по мере прогрессирования ХБП. Интимальная кальцификация коронарных артерий является фактором риска ОИМ [1, 19]. У больных ХБП кальцификация медии имеет протяженный диффузный характер (артериосклероз Менкеберга), ведущий к поражению коронарных артерий
с разитием острого коронарного синдрома, эктопической кальцификации крупных артерий, что вызывает повышение скорости пульсовой волны, клинически проявляющейся увеличением систолического и пульсового давления и быстрым формированием ГЛЖ [21–24].
Кальцификация сердца проявляется главным образом кальцинозом коронарных артерий и клапанов, клиническим проявлением которого могут быть митральная недостаточность или стеноз, нарушения сердечного ритма, ХСН, но может быть представленной и диффузной кальцификацией миокарда [1, 19].
Факторы риска кальцификации сосудов:
• увеличение концентрации кальция и фосфора в крови;
• произведение Са2+ × Р5+ более 55 мг2/дл2 (5,5 ммоль2/л2);
• увеличение концентрации иПТГ в крови;
• дефицит белка фетуина А.
Кальцификация сосудов может затруднять формирование артериовенозной фистулы и создавать дополнительные трудности при пересадке почки.
Кальцификации могут подвергаться артерии предплечья, запястья, кистей, нижних конечностей, брюшной полости, грудной клетки, таза, головного мозга [1, 2, 18].
У некоторых больных кальцификация сосудов бывает столь выраженной, что сосуды конечностей невозможно пережать манжеткой для измерения артериального давления [1, 19].
Для диагностики и измерения содержания кальция в сосудистой стенке используют электронно-лучевую томографию [1, 20, 23].
Механизмы внекостной кальцификации при ХБП имеют сложный и многофакторный характер. Продемонстрировано, что VSMC и другие клетки сосудов (перициты, фибробласты) могут трансформироваться в клетки, подобные остеобластам, и продуцировать гидроксиапатит – основное минеральное вещество кости. В условиях гиперфосфатемии гладкомышечные клетки сосудов способны аккумулировать фосфор, экспрессировать гены костных белков и становиться очагами
кальцификации [20, 25].
ВГПТ у пациентов ХБП может вносить вклад в высокую степень выраженности сердечно-сосудистых осложнений, участвуя в развитии миокардиального фиброза. Установлено, что иПТГ не только повышает клеточную нагрузку Са2+, усугубляя атеросклеротические изменения, но и активирует
фибробласты, а также обладает прямым повреждающим действием на миокард за счет нарушения метаболизма кардиомиоцитов [1, 19, 25].
Миокардиальный фиброз, кальцификация сосудов сердца и клапанного аппарата способствуют развитию и прогрессированию систолической дисфункции, возникновению ХСН. ГЛЖ и повышение содержания Са2+ в кардиомиоцитах приводят к нарушению и диастолической функции миокарда. Эти нарушения – главная причина сердечно-сосудистой смертности пациентов с ХБП [19–21].
Важными медиаторами взаимодействия “остеогенез–ангиогенез” являются индуцируемый гипоксией фактор1-α (HIF1-α) и сосудистый эндотелиальный фактор роста (VEGF), которые, как предполагают, с помощью ангиогенных сигналов индуцируют сосудистую кальцификацию [19, 20].
Начальный этап кальцификации медии артерий связан с деградацией эластина. Установлено, что матриксные металлопротеиназы (ММР-2 и ММР-9) разрушают эластин, а образовавшиеся при этом растворимые эластин-пептиды связываются с рецепторами ламинин-эластина (ELR), расположенными на поверхности гладкомышечных клеток. В разрушении эластина участвует и избыточно экспрессируемый трансформирующий фактор роста β (TGF-β). Известно, что он также усиливает кальцификацию VSMC и играет важную роль в дифференциации остеобластов. Остеогенная трансформация VSMC активируется последовательными сигналами от ELR, TGF-β, митоген-активируемой протеинкиназы (MAPK). При кальцификации медии экспрессируется ряд белков, связанных с остеогенезом: остеокальцин, остеопонтин, матриксный γ-GLA (carboxyglutamic acid), протеин (MGP) и остеопротегрин [1, 18–20, 24].
Таблица 2. Связь повышения плазменной концентрации FGF-23 с неблагоприятным прогнозом больных ХБП (данные проспективных исследований).
Связь остеогенеза с эктопической минерализацией. Установлена тесная связь между процессами остеогенеза и кальцификацией сосудов. В последние годы были идентифицированы ранее
неизвестные факторы, продуцируемые почками и костью, участвующие в регуляции гомеостаза фосфата, витамина D и минерализации костной ткани, морфогенетические белки (FGF-23 и Klotho). Уровень FGF-23 (фактора роста фибробластов 23) – белка, продуцируемого преимущественно
остеоцитами, заметно увеличивается уже в преддиализных стадиях ХБП [18, 26–30].
Функциональная роль повышения в циркуляции FGF-23 при различных стадиях ХБП в последнее время активно изучается.
Установлено, что на 2–3-й стадиях ХБП повышение продукции остеоцитами FGF-23 вносит свой вклад в адаптивное увеличение экскреции фосфора и снижение продукции кальцитриола [31–37].
В дальнейшем при снижении СКФ на 4–5-й стадиях ХБП увеличение FGF-23 не может предотвращать развитие гиперфосфатемии и формирование ВГПТ. Данные проспективных исследований [27–30] свидетельствуют, что повышение уровня FGF-23 у больных ХБП на момент начала ГД ассоциировано
со смертностью и сосудистой кальцификацией независимо от установленных факторов риска и уровня фосфора и иПТГ в сыворотке, т. е. FGF-23 является потенциальным независимым “уремическим токсином” (табл. 2).
Белок Klotho существует в двух формах – трансмембранной и экстрацеллюлярной (секретируемой). Он продуцируется и секретируется клетками проксимальных почечных канальцев, трансмембранная форма продуцируется также клетками ПЩЖ [38–39].
В экспериментальном исследовании K. Makoto было установлено, что трансмембранная форма белка Klotho является корецептором для FGF-23 и в этом качестве участвует в регуляции обмена фосфора, кальция и витамина D.Экстрацеллюлярная часть белка Klotho, секретируемая в кровоток, функционирует как эндокринный фактор и кодирует многие факторы роста, в т. ч. инсулиноподобный фактор роста-1 и Wnt [39].
Экспрессия белка Klotho снижена у больных ХБП, на основании чего предполагают, что Klotho является ренопротективным фактором. Становится все более очевидным, что регуляция метаболизма фосфора играет важную роль в патогенезе ХБП и что для улучшения качества жизни пациентов с ХБП гиперфосфатемию необходимо рано корригировать, влияя на эндокринные взаимодействия “кости–почки–паращитовидные железы”, опосредованные Klotho и FGF-23 [26–37].
Поскольку уровень сывороточного FGF-23 у больных ХБП предшествует гиперфосфатемии, резистентность к FGF-23 может быть одним из наиболее ранних проявлений нарушения метаболизма фосфора при ХБП. Предполагают, что развитие резистентности к FGF-23 вызвано снижением почечной экспрессии Klotho. В связи с этим низкий уровень экспрессии Klotho в почках может быть фактором неблагоприятного
отдаленного прогноза диализных больных [39].
Так как концентрация FGF-23 снижается при уменьшении гиперфосфатемии, терапевтической стратегией у таких больных должны быть диета с низким содержанием фосфатов и применение фосфат-биндеров уже с ранней стадии ХБП. Эти меры могут способствовать профилактике сердечно-сосудистых осложнений и развития ВГПТ у больных ХБП. В то же время заслуживает внимания тот факт, что у пациентов с ХБП уровень FGF-23 уже натощак повышен, притом часто в отсутствие гиперфосфатемии и гипокальциемии. Хотя в настоящее время точно не установлено, на какой стадии ХБП начинает увеличиваться сывороточный уровень FGF-23, можно предположить, что увеличение продукции FGF-23 предшествует повышению содержания в крови фосфора и иПТГ [26–29].
В большинстве клинических исследований измерение уровней фосфора, кальция, FGF-23 и иПТГ в крови проведено натощак. В то же время у больных на ранней стадии ХБП без гиперфосфатемии и гипокальциемии после приема пищи содержание FGF-23 и иПТГ увеличивается. Так, у 13 пациентов с ХБП с нормальным уровнем фосфора и кальция натощак и 21 здорового лица после еды экскреция фосфора с мочой увеличивалась, несмотря на нормальные уровни фосфора и FGF-23 в крови. Экскреция кальция с мочой после еды также увеличивалась в обеих группах, но только у лиц с ХБП это сопровождалось существенным снижением кальция и повышением уровня иПТГ в крови. Следовательно, FGF-23 не оказывает влияния на степень фосфатурии после приема пищи как у здоровых лиц, так и у больных ХБП, однако наблюдаемая после еды у больных ХБП транзиторная каль-
циурия, сопровождающаяся относительной гипокальциемией, может быть отражением раннего механизма формирования ВГПТ [16, 26].
Результаты ряда исследований последних лет позволяют считать, что измерение уровня иПТГ натощак может быть малоинформативным для выявления ВГПТ в ранней его стадии и что, возможно, определение плазменной концентрации FGF-23 или измерение его уровня после еды может оказаться более чувствительным скрининговым тестом для диагностики ВГПТ [27, 28]. FGF-23 определяют в сыворотке крови с помощью набора FGF-23 Elisa Kit. Klotho диагностируют в сыворотке крови, спинномозговой жидкости, моче с помощью набора анти-Klotho поликлональных антител. Методом иммуногистохимического анализа возможно определение FGF-23/Klotho в проксимальных канальцах почек [26, 29].
Таким образом, нарушение у больных ХБП фосфорно-кальциевого обмена с развитием гиперфосфатемии, сосудистой кальцификации, ГЛЖ, опосредуемых в т. ч. вновь открытыми механизмами регуляции – Klotho и FGF-23, является важным фактором риска сердечно-сосудистых осложнений – одной из причин высокой смертности этой категории больных.
Ингибиторы эктопической минерализации. Важную роль в механизме кальцификации артерий играет дефицит ингибиторов кальцификации.
Матриксный γ-карбоксиглютаровокислый протеин (Matrix γ-Carboxyglutamic Acid Protein) – MGP – белок, продуцируемый костью. MGP блокирует остеохондроцитарную трансдифференциацию сосудистых гладкомышечных клеток в остеобластоподобные клетки. К тому же MGP препятствует
образованию ядер кристаллизации в медии и атеросклеротических бляшках, является регулятором синтеза скелетного матрикса [1, 16, 18].
Остеопонтин – кислый фосфопротеин, экспрессируется в очагах кальцификации артерий, является активатором остеобластов и ингибитором образования гидроксиапатита. Проникая в очаги кристаллизации, остеопонтин ингибирует их образование [1, 18].
Остеопротегрин – белок, экспрессируется в атеросклеротических бляшках и очагах кальцификации медии. Остеопротегрин ингибирует дифференциацию остеокластов и активность ЩФ, тем самым замедляет кальцификацию медии и атеросклеротических бляшек [18].
Фетуин-А (протеин α2 Heremans-Schmid) – кальцийсвязывающий белок, продуцируемый преимущественно в печени, содержание которого существенно снижается при уремии. В экспериментальных исследованиях связывание фетуина А приводило к развитию кальцификации сосудов. Фетуин А, депонируясь в сосудистых гладкомышечных клетках, ингибирует их превращение в остеобластоподобные клетки [18].
Пирофосфаты подавляют остеохондрогенную трансдифференциацию сосудистых гладкомышечных клеток и образование кристаллов гидроксиапатита. Синтезируются из нуклеотидтрифосфатов при участии фермента нуклеотидпирофосфатазы. При ХБП содержание нуклеотидпирофосфатазы и переносчика пирофосфатов в кальцифицирующихся слоях медии снижено. Ингибируют дифференциацию сосудистых гладкомышечных клеток в остеобластоподобные клетки и кальцификацию медии и атеросклеротических бляшек также IGF-1- и N-3-жирные кислоты [1, 16, 18].
Стимуляторы эктопической минерализации. Гиперфосфатемия вызывает индукцию факторов остеобластной дифференциации, таких как Cbfa1/Runx2, остеокальцин и натрийзависимый
котранспортер фосфатов III типа – Pit-1. Экспрессия Pit-1 стимулируется также высокой концентрацией внеклеточного кальция. В окружающее внеклеточное пространство сосудистой
стенки начинают секретироваться матриксные пузырьки, белки костного матрикса, которые в последующем подвергаются минерализации [1, 18].
Уремические токсины вызывают экспрессию Cbfa1/Runx2, остеопонтина, а также ЩФ и остеопротегрина вне зависимости от концентрации фосфора. Они увеличивают секрецию
белка костного морфогенеза (BMP-2) сосудистыми гладкомышечными клетками, ускоряя их минерализацию. По мере прогрессирования почечной недостаточности в механизм формирования кальцификации артерий включаются новые факторы [1, 40, 41].
Оксидативный стресс и воспаление. Уремическая интоксикация сопряжена с оксидативным стрессом и синдромом системного воспаления. Последний в свою очередь осложняет свойственный уремии оксидативный стресс. Образование оксидантов – еще один из факторов, стимулирующих повышенную окисляемость липопротеинов, которые ускоряют трансформацию сосудистых гладкомышечных клеток в остеобластоподобные клетки. Остеобластную трансформацию VSMC стимулируют также ГКС, лептин и гипергликемия [1, 18].
Роль нарушений метаболизма витамина D. Получены данные об участии витамина D в регулировании активности и метаболизма VSMC. Так, установлено, что сосудистые гладкомышечные клетки экспрессируют 1α-гидроксилазу для превращения 25(OH)D3 в 1,25(OH)2D3 и рецепторы к витамину
D (VDR). Кальцитриол усиливает экспрессию на гладкомышечных клетках VDR – фактора клеточной пролиферации и дифференциации гладкомышечных клеток. В зависимости от концентрации в крови кальцитриол может как стимулировать, так и подавлять пролиферацию сосудистых гладкомышечных клеток. Так, кальцитриол в концентрациях 10-7–10-10 М дозозависимо стимулирует кальцификацию сосудов за счет повышения соотношения "активатор рецептора нуклеарного фактора/остеопротегрин". В экспериментальных моделях ХБП аналоги кальцитриола (парикальцитол и доксекальциферол) в меньшей степени, чем кальцитриол в сопоставимых дозах, вызывали кальцификацию сосудов. Парикальцитол в отличие от кальцитриола не приводил к нарастанию экспрессии Cbfa1/Runx2 и остеокальцина. В дозах, достаточных для подавления гиперпродукции иПТГ, кальцитриол и его аналоги защищали от кальцификации сосуды, но более высокие дозы препаратов
стимулировали кальцификацию [1, 16, 18, 19].
В исследовании (медиана продолжительности наблюдения – 21 год), включившем 520 больных ХБП со средней СКФ 30 мл/мин/1,73 м2, лечение кальцитриолом способствовало снижению летальности. Сопоставимые результаты получены и в другом аналогичном исследовании [1].
Таким образом, исследования последних лет свидетельствуют, что у пациентов с ХБП кальцитриол и его аналоги в высоких дозах могут стимулировать кальцификацию сосудов, а в умеренных — оказывать протективное действие путем ингибирования продукции коллагена I типа и промоутера гена core-binding factor-α1 (Cbfa1). Коллаген I типа является основой для депозитов кальция, а Cbfa1 стимулирует образование коллагена I типа [40, 41].
Клинические проявления кальцификации мягких тканей. Метастатическая кальцификация бывает при ВГПТ и у больных адинамическим заболеванием скелета, склонных к гиперкальциемии из-за неспособности костной ткани усваивать избыток кальция. Гиперфосфатемия с повышением произведения Са2+ × Р5+ > 55 мг2/дл2 (4,5–5,5 ммоль2/л2), алкалоз, развивающийся во время ГД, локальное повреждение тканей предрасполагают к отложению фосфата кальция в мягких тканях [1, 16, 18].
Установлено, что кальцификаты мягких тканей и сосудов состоят из кристаллов гидроксиапатита с соотношением Са : Мg : Р, идентичным таковому в кости, тогда как в мышцах, сердце и легких обычно находят аморфные микрокристаллы солей кальция, магния и фосфатов – СаМg3(РО4)2. Предполагают,
что различный состав кальцификатов обусловлен местными тканевыми факторами – концентрацией водородных ионов, магния, кальция и фосфора. Выпадение кристаллов гидроксиапатита сопровождается выраженной фиброзной реакцией, а аморфные кристаллы ее не вызывают [1, 2].
Отложение фосфорно-кальциевых солей в коже вызывает сильнейший зуд, исчезающий только после паратиреоидэктомии [1, 18, 19].
Кальцификация кожи. Депозиты солей кальция в коже характеризуются образованием макул (пятен) или папул (узелков), состоящих из твердых образований, в которых при биопсии выявляют кристаллы солей кальция. После субтотальной паратиреоидэктомии депозиты солей кальция в коже регрессируют, что свидетельствует о ведущей роли ВГПТ в генезе кальцификации кожи [16, 18].
Образование кожных язв и некроз тканей (кальцифилаксия). Синдром впервые описан Seyle в 1962 г. и назван автором кальцифилаксией. В последние годы многие авторы используют другое название этого синдрома – кальцифицирующая уремическая артериопатия [1]. Это клинический синдром,
характеризующийся развитием прогрессирующего ишемического поражения кожи с вовлечением пальцев рук и ног, бедер и голеней, может развиваться у пациентов, получавших лечение регулярным ГД в течение 10 и более лет. Менее часто он развивается у пациентов на ПАПД. У больных почти всегда также обнаруживают кальцификацию сосудов и рентгенологические признаки субпериостальной резорбции кости. Перед появлением язв или некрозов тканей обычно развиваются
болезненные эритематозные подкожные узелки или синеватые пятна. Повреждениям пальцев кисти и стоп может предшествовать и синдром Рейно. Язвы могут развиваться медленно (в течение нескольких месяцев) или быстро – за несколько недель. Присоединение инфекции часто приводит к сепсису
со смертельным исходом [1, 18].
Тактика ведения таких больных окончательно не определена. Местное лечение не эффективно, субтотальная паратиреои эктомия у большинства больных приводила к регрессу синдрома кальцифилаксии, у части больных повреждения кожи не заживали, а у некоторых они даже усугублялись.
Нарушения минерального обмена с развитием ВГПТ и сосудистой кальцификации, локальное повреждение тканей, ожирение, особенно у женщин белой расы, дефицит протеина С предрасполагают к развитию данного синдрома. Имеются сообщения, что развивающийся дефицит протеина С у пациентов с ХБП может приводить к гиперкоагуляции и, следовательно, к окклюзии сосудов и некрозу тканей, однако применение варфарина у таких больных сопровождается
усугублением кожных язв. Локальное повреждение кожи в месте инъекций инсулина, гепарина или декстрана железа нередко становится местом, где образуются язвы и участки некроза кожи [1, 16].
Кальцификация глаз. Депозиты солей кальция в конъюнктиве и роговице на фоне сосудистого воспаления обусловливают “красные глаза”. Это транзиторный процесс, который рецидивирует каждый раз, когда происходят новые отложения кальция в конъюнктиву [1].
Конъюнктивальные отложения солей кальция могут быть бессимптомными и выявляться только при исследовании с помощью щелевой лампы.Отложения солей кальция видны как белые бляшки или как мелкие точечные депозиты на латеральном или медиальном сегменте конъюнктивы глазного яблока. Кальциевые депозиты также могут локализовываться в области роговицы на латеральном или медиальных сегменте каймы глаз (т. н. ленточная кератопатия). Отложению депозитов кальция в сосудах глаз способствует увеличиние локального рН тканевой жидкости вследствие потери СО2 через конъюнктивальную поверхность глаз [18].
Висцеральная кальцификация. Чаще всего отложения солей кальция обнаруживаются в легких, желудке, миокарде, скелетных мышцах и почках. Эти кальцификаты обычно не выявляются при рентгенологическом исследовании, но могут быть обнаружены с помощью изотопного сканирования с
99mTc-пирофосфатом. Кальцификаты в легких и мышце сердца могут вести к тяжелым и даже смертельным осложнениям. Описаны случаи, когда кальцификаты в легких приводили к тяжелому легочному фиброзу, легочной гипертензии и ГЛЖ. Кальцификаты в легких и мышце сердца являются одной из главных причин увеличенной заболеваемости и смертности диализных больных [1, 16, 18, 42].
Избыточное содержание в рационе витамина С, метаболизирующегося в щавелевую кислоту, может привести к отложению щавелевокислого кальция в мягких тканях, миокарде, митральных и аортальных клапанах с развитием кардиомиопатии и ХСН со смертельным исходом на диализе [1].
Околосуставная кальцификация. Отложение солей кальция около суставов ограничивает их подвижность и сопровождается тендовагинитом. Синовиальная жидкость вовлеченных суставов – прозрачная с нормальной вязкостью и нормальным количеством клеток [1, 43, 44]. Это острое околосуставное заболевание называют кальцифицирующим периартритом. Помимо солей кальция в околосуставных тканях могут откладываться кристаллы мочевой кислоты (вторичная подагра), что нередко сочетается с хондрокальцинозом. Псевдоподагру диагностируют при обнаружении в синовиальных клетках кристаллов пирофосфата [2]. В исследовании, включившем 135 больных на ГД, распространенность околосуставной кальцификации увеличилась с 9 до 42 % с первого по восьмой год диализного лечения [18].
Иногда у диализных больных рядом с суставами образуются крупные опухолеподобные массы, состоящие из инкапсулированной известковой жидкости и пастообразных солей кальция. Эти образования обычно бывают безболезненными, но могут ограничивать подвижность суставов за счет своего размера. Опухолеподобные депозиты кальция часто регрессируют при адекватном контроле уровней сывороточного фосфора применением фосфат-связывающих препаратов или после
субтотальной паратиреоидэктомии [1, 16, 18].
Профилактика и лечение нарушений фосфорно-кальциевого обмена при ХБП
Диета и фосфат-биндеры
Низкофосфорная диета и/или применение фосфат-биндеров все чаще признаются в качестве важного терапевтического подхода к предотвращению опасных для жизни осложнений у больных ХБП. Согласно Клиническим практическим рекомендациям K/DOQI, потребление фосфора следует ограничивать до 800–1000 мг/сут (с коррекцией на пищевую потребность в белке), если уровень фосфора в сыворотке крови выше 4,6 мг/дл (1,49 ммоль/л) при ХБП III–IV стадий и выше 5,5 мг/дл (1,78 ммоль/л) у диализных больных или содержание иПТГ в плазме превышает целевой уровень, определенный для соответствующей стадии ХБП [1, 16]. Если, несмотря на ограничение потребления фосфатов с пищей не удается контролировать уровень фосфора или иПТГ в пределах целевых значений, необходимо назначать фосфат-биндеры [19, 20]. В России для коррекции гиперфосфатемии наиболее часто используют кальция карбонат и кальция ацетат. Способность связывать фосфор у кальция ацетата в 2 раза выше, чем у кальция карбоната [1, 16]. Длительный прием фосфат-связывающих препаратов на основе солей кальция может вызывать гиперкальциемию, которая на фоне приема кальция карбоната бывает в 3,5 раза чаще, чем при использовании кальция ацетата, т. к. взаимодействие кальция карбоната с фосфором начинается при рН 5,0, когда растворимость
кальция карбоната снижается [1, 18, 19]. Фосфат-биндеры на основе кальция эффективно снижают концентрацию фосфора в сыворотке и могут использоваться в качестве начальной фосфат-связывающей терапии. При этом суммарная доза элементарного кальция, используемого для связывания фосфора, поступающего с пищей, не должна превышать 1,5 г/сут [1, 16]. Фосфат-биндеры на основе кальция не должны применяться у диализных больных с гиперкальциемией (корректированный общий кальций сыворотки – выше 10,2 мг/дл [2,54 ммоль/л]) и в тех случаях, когда уровень ПТГ плазмы ниже 150 пг/мл (16,5 пмоль/л) при 2 последовательных измерениях [1]. У таких больных следует отдавать предпочтение фосфат-биндерам, не содержащим кальций [1, 2, 16]. В настоящее время в клинической практике для связывания в ЖКТ фосфора, поступающего с пищей, все более широкое применение находит севеламера гидрохлорид (ренагель) — синтетический препарат, не содержащий ни кальций, ни гидроокись алюминия. Препарат не абсорбируется в ЖКТ, снижает в крови содержание иПТГ, фосфора и произведение Са × Р, частично корригирует дислипидемию [1, 16]. В связи с отсутствием кальциемического эффекта отмечается безопасность комбинации ренагеля с активными аналогами витамина D, а также его эффективность при кальцификации сосудов и мягких тканей. Показанием к назначению ренагеля считают повышение сывороточных уровней фосфора и
корректированного общего кальция выше 5,5 и 10,2 мг/дл соответственно при снижении иПТГ ниже 150 пг/мл и развитии метастатической кальцификации [1, 16]. При длительном применении ренагеля может снижаться диуретический эффект фуросемида, развиваться гипохлоремический ацидоз, связанный с потерей бикарбоната [1, 16, 45, 46]. Выбор дозы севеламера гидрохлорида в зависимости от исходного уровня фосфора в сыворотке крови представлен в табл. 3.

Переход с фосфат-биндеров на основе кальция на севеламера гидрохлорид осуществляют в эквивалентных дозах, рассчитанных в мг/кг, сравнительно с препаратами на основе кальция.
Доза севеламера гидрохлорида может варьироваться от 1 до 5 таблеток по 800 мг за один прием пищи [1]. В США и Европе в целях связывания в ЖКТ фосфора используют лантана карбонат (Fosrenol). Препарат производится в таблетках по 500, 750 и 1000 мг. Начальная доза – 750 мг ежедневно во время или сразу после еды. Продолжительность лечения – до нормализации уровня фосфора (1,13–1,78 ммоль/л; 3,5–5,5 мг/дл). Максимальная доза препарата – 3750 мг/сут (на короткий период времени – 5–7 дней). Однако лантан частично всасывается в кровоток и может аккумулироваться в костной ткани [1, 16]. В настоящее время проводят клинические испытания севеламера карбонат и Zerenex-фосфат-биндера, содержащего соединения неорганического железа. Предварительные данные свидетельствуют о том, что их эффективность связывать в ЖКТ фосфор не уступает солям кальция [1, 16, 47].
У больных на ЗПТ увеличить элиминацию фосфора можно путем замены мембран из целлюлозы ацетата на полисульфоновые мембраны либо использовать гемодиафильтрацию, при которой клиренс фосфора выше, чем во время ГД. Увеличение скорости кровотока через гемодиализатор, хотя и увеличивает клиренс креатинина, не влияет на выведение фосфора, в то время как увеличение времени диализа (например, перевод больных с 3 раз в неделю на ежедневный ГД) на 30 % снижает
преддиализный уровень фофора в сыворотке крови и позволяет уменьшать частоту приема фосфат-связывающих препаратов [1, 16]. У больных на ПД клиренс креатинина и фосфора повышается при увеличении частоты смен диализирующего раствора в брюшной полости. Уровень корректированного
общего кальция сыворотки крови следует поддерживать в пределах нормального диапазона (8,4–9,5 мг/дл [2,1–2,37 ммоль/л]) [1]. У диализных больных, получающих фосфатбиндеры на основе кальция и активные метаболиты витамина D ри достижении уровня корректированного общего кальция
сыворотки крови выше 10,2 мг/дл [2,54 ммоль/л], следует уменьшать дозы препаратов или совсем прекращать лечение до возвращения уровня корректированного общего кальция сыворотки к целевым значениям. При безуспешности этих мер следует использовать диализат с низким содержанием кальция (1,5–2,0 мэкв/л) в течение 3–4 недель. Фосфорно-кальциевое произведение необходимо поддерживать на уровне ниже 55 мг2/дл2, что лучше всего достигается за счет контроля
уровня фосфора сыворотки крови в пределах целевого диапазона [1, 16, 19, 47].
Таблица 4. Сывороточные уровни иПТГ, корректированного общего кальция и фосфора, при которых показаны активные метаболиты витамина D. Начальные дозы препарата у больных ХБП и III IV стадий.
С помощью коррекции гиперфосфатемии может быть снижена концентрация FGF-23 у пациентов с ХБП. Поскольку повышение сывороточного уровня FGF-23 связано с увеличением смертности пациентов с терминальной стадией ХБП, в последнее время для коррекции избыточной продукции FGF-23, кроме того, обсуждается перспектива применения анти-FGF-23 нейтрализующих антител, однако, насколько
это будет клинически полезным, пока неясно [48].
Лечение гипокальциемии должно включать назначение солей кальция, например карбоната кальция и/или метаболитов витамина D внутрь [1, 16].
Лечение активными метаболитами витамина D
Если плазменный уровень иПТГ выше целевого диапазона для данной стадии ХБП, а сывороточный уровень 25(ОН)D3 ниже 30 нг/мл, следует начинать лечение витамином D2 (эргокальциферол). Считают, что эргокальциферол на ранних стадиях ХБП более безопасен, чем кальциферол [1, 16,
47]. Дозу препарата необходимо выбирать, ориентируясь на выраженность дефицита 25(ОН)D3. При уровне 25(ОН)D3 ниже 15 нг/мл (37 нмоль/л) в сыворотке крови эффективна доза эргокальциферола 50 000 МЕ/нед. × 4 недели с последующим переходом на режим 50 000 МЕ/мес. × 4 месяца. Если сывороточный уровень 25(ОН)D3 равен 20–30 нг/мл (50–75 нмоль/л), препарат следует назначать в дозе 50 000 МЕ ежемесячно в течение 6 месяцев [1, 16]. Лечение витамином D также показано у больных на додиализных стадиях ХБП с нормальным содержанием 25(ОН)D3 (выше 30 нг/мл [75 нмоль/л]), но сниженным уровнем корректированного общего кальция и повышенным иПТГ в сыворотке крови при нормальных значениях сывороточного уровня фосфора. Для подавления синтеза и секреции иПТГ более эффективны активные метаболиты витамина D (кальцитриол, альфакальцидол или доксекальциферол) [1, 16, 19, 47]. Начальные дозы препаратов должны быть низкими (табл. 4).
Доза кальцитриола не должна превышать 0,5 мкг/сут, за исключением случаев, когда корректированный уровень общего кальция увеличивается менее чем на 0,2–0,3 мг/дл/мес. [1, 16]. Лечение активными метаболитами витамина D следует предпринимать только у больных с сывороточными уровнями корректированного общего кальция ниже 9,5 мг/дл (2,37 ммоль/л) и сывороточного фосфора ниже 4,6 мг/дл (1,49 ммоль/л) [1, 18]. Больные, получающие лечение ГД или ПД, у которых сывороточные уровни иПТГ выше 300 пг/мл (33,0пмоль/л), должны принимать активные метаболиты витамина D (такие как кальцитриол, альфакальцидол, доксекальциферол) или селективный активатор ВДР парикальцитол (табл. 5), чтобы уменьшить сывороточную концентрацию иПТГ до целевого уровня 150–300 пг/мл (16,5–33,0 пмоль/л) [1, 16, 19, 47].
Больным на ПД назначают кальцитриол или доксекальциферол внутрь в дозе 0,5–1,0 и 2,5–5,0 мкг 2–3 раза в неделю соответственно [1, 16]. В качестве альтернативы можно рекомендовать ежедневно принимать кальцитриол внутрь в дозе 0,25 мкг [19]. В начале лечения активными метаболитами
витамина D или при увеличении дозы принимаемых препаратов сывороточные уровни кальция и фосфора должны мониторироваться каждые 2 недели в течение первого месяца, затем 1 раз в месяц [1, 16]. Гиперплазия ПЩЖ исчезает у большинства больных после эффективного подавления секреции
иПТГ [1]. Высокие концентрации 1,25(OH)2D3 стимулируют апоптоз клеток ПЩЖ [1, 16]. Этот феномен используется для медикаментозной “паратиреоидэктомии” – впрыскивания 1,25(OH)2D3 непосредственно в гиперплазированные паращитовидные железы [1, 47]. Для супрессии иПТГ перспективно применение парикальцитола – 1,25(ОН)2D2 – селективного активатора рецепторов к витамину D, в структуре которого имеется модификация боковой цепи (D2) и кольца А (19-нор).
Парикальцитол (Земплар) селективно индуцирует экспрессию гена VDR (S-VDRA) на ПЩЖ, подавляет секрецию иПТГ, не активирует VDR в кишечнике и почти не влияет на резорбцию костной ткани, поэтому реже вызывает гиперкальциемию, чем активные метаболиты витамина D. Препарат выпускают в ампулах по 1 мл (5 мкг) и в капсулах по 1, 2 и 4 мкг. У пациентов на ЗПТ препарат можно вводить медленно внутривенно в течение не менее 30 секунд, чтобы свести к минимуму боль при инфузии. Рекомендуемая стартовая доза парикальцитола составляет 0,04–0,10 мкг/кг. Выбор дозы парикальцитола зависит от исходного уровня иПТГ: стартовая доза (мкг) =иПТГ (пг/мл) / 80. Ее вводят в виде болюса не чаще, чем через день во время диализа. Парикальцитол в капсулах назначается
один раз в день, ежедневно или 3 раза в неделю [1, 16]. Данные, основанные на трех проспективных рандомизированных многоцентровых исследованиях, свидетельствуют об эффективном подавлении парикальцитолом секреции иПТГ, а также снижении активности костного изофермента ЩФ и содержания остеокальцина в сыворотке крови, свидетельствующих об уменьшении костной резорбции [1].
Таблица 5. Рекомендуемые начальные дозы активных метаболитов витамина D и парикальцитола в зависимости от сывороточных уровней иПТГ, кальция, фосфора и произведения Ca x P у больных на ГД.
У больных ХБП 3–4-й стадий, у которых плазменные уровни иПТГ выше 70 пг/мл (7,7 пмоль/л) и 110 пг/мл (12,1 пмоль/л) соответственно, кроме мероприятий, ограничивающих потребление фосфора с пищей, в целях предупреждения или лечения костной патологии следует назначать кальцитриол
или альфакальцидол либо доксекальциферол – табл. 5 [1]. Больным ХБП 5-й стадий, имеющим повышение уровня иПТГ более 300 пг/мл (33,0 пмоль/л) и при наличии симптомов ренальной остеодистрофии для подавления избыточной секреции иПТГ и нормализации повышенного метаболического профиля костной ткани показано назначение кальцитриола или доксекальциферола, альфакальцидола и парикальцитола [1, 16, 19, 49, 50].
Таким образом, нарушение метаболизма фосфора играет важную роль в патогенезе ХБП; для улучшения качества жизни пациентов с ХБП гиперфосфатемию необходимо рано корригировать. Механизмы внекостной кальцификации при ХБП имеют сложный и многофакторный характер. В последние годы идентифицированы медиаторы взаимодействия “остеогенез–ангиогенез”, индуцируемый гипоксией фактор1-α (HIF1-α) и сосудистый эндотелиальный фактор роста (VEGF), которые с помощью ангиогенных сигналов индуцируют сосудистую кальцификацию. Определены ранее не известные факторы, продуцируемые почками и костью, участвующие в регуляции гомеостаза фосфора, витамина D и минерализации костной ткани, – морфогенетические белки (FGF-23 и Klotho). Предполагают, что одним из наиболее ранних проявлений нарушения метаболизма фосфора при
ХБП является резистентность к FGF-23, обусловленная снижением почечной экспрессии Klotho. В эксперименте получены данные о том, что парикальцитол увеличивает экспрессию Кlothо в почках. Получены и такие данные: кальцитриол и парикальцитол увеличивают экспрессию Кlothо в почках. В последнее время для коррекции избыточной продукции FGF-23 обсуждается перспектива применения анти-FGF-23 нейтрализующих антител, однако в условиях сложного взаимодействия “кости–почки–паращитовидные железы” их применение требует дальнейшего изучения.



{kind=link}
{kind=link}
{kind=link}