
Введение
Болезнь Кастлемана (БК) представляет собой гетерогенную группу лимфопролиферативных заболеваний с характерными общими морфологическими признаками при биопсии л/у. Первое описание клинического случая локального опухолевидного образования в переднем средостении без системных проявлений у 41-летнего мужчины принадлежит B. Castleman и V. Towne (1954). Гистологическая картина опухоли не укладывалась ни в одну из известных в то время нозологий и характеризовалась увеличением числа лимфоидных фолликулов, гиалинозом в зародышевых центрах фолликулов, выраженной пролиферацией капилляров, включая фолликулярную и межфолликулярную гиперплазию эндотелия [1]. Через 2 года B. Castleman et al. [2] опубликовали серию 12 наблюдений с более подробным описанием нескольких типов гистологической картины в л/у, получивших название в честь первооткрывателя «болезнь Кастлемана».
С момента первоначального описания БК достижения в диагностике, классификации, патогенезе и терапии являются столь существенными, что в 2018 г. основан Всемирный день болезни Кастлемана, который проводится ежегодно 23 июля.
Классификация
В зависимости от клинической картины и течения заболевания, патологических, вирусологических особенностей современная классификация БК включает уницентрический (одноцентрический) вариант (УБК) с локализованным поражением одиночного л/у и генерализованный или мультицентрический (МБК) вариант с лимфаденопатией в нескольких узлах (рис. 1).
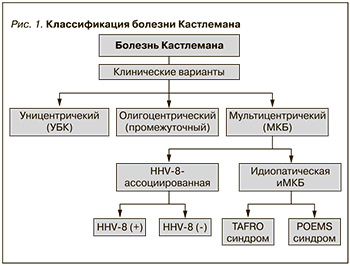
Мультицентрический вариант БК дополнительно включает подтипы, связанные с вирусом герпеса человека 8 (HHV8), также известного, как вирус саркомы Капоши (HHV8-МКБ), и идиопатический вариант (иМБК), который является HHV8-негативным. HHV8-МКБ может возникать как у людей, живущих с ВИЧ, так и у лиц с ослабленным иммунитетом по другой причине. HHV8-МКБ, встречающийся при ВИЧ-инфекции, может быть обнаружен в сочетании с другими новообразованиями, включая саркому Капоши, В-клеточные лимфомы и лимфому Ходжкина. Важно отметить, что эти злокачественные новообразования постоянно ассоциируются с саркомой Капоши и первичной лимфомой и часто связаны с инфекцией, вызванной вирусом Эпштейна–Барр.
Идиопатический вариант МБК также неоднороден по своему составу и может сопровождаться такими тяжелыми клиническими синдромами, как TAFRO (тромбоцитопения, асцит, лихорадка, ретикулярный фиброз костного мозга, почечная дисфункция и органомегалия) или POEMS (полиневропатия, органомегалия, эндокринопатия, наличие моноклональной секреции и изменения кожи и ее придатков) [3–7].
Совсем недавно был описан промежуточный вариант БК – олигоцентрический, или региональный, при котором поражаются л/у двух-трех соседних областей. Считают, что клиническое течение промежуточного варианта БК аналогично УБК, однако клинические и лабораторные отклонения не в полном объеме соответствуют диагностическим критериям МБК [8].
Важно подчеркнуть, что все варианты БК демонстрируют ряд гистопатологических особенностей, которые исторически классифицируются на гиалино-васкулярные, плазмоклеточные (плазмоцитарные и/или плазмобластические) или смешанно-клеточные (промежуточные) варианты [9–11].
Заболеваемость
Следует отметить, что существуют ограниченные данные о распространенности БК в разных странах. Из-за отсутствия согласованных диагностических критериев оценка частоты БК до 2017 г. основывалась на описании отдельных наблюдений или серии клинических случаев. В 2017 г. были разработаны руководящие принципы, которые включали диагностические критерии и классификацию БК [12, 13].
Эпидемиология УБК заметно отличается от эпидемиологии МБК. Так, в США ежегодно регистрируют примерно 6500–7000 случаев БК, при этом УБК составляет 75%, что соответствует 16 случаям на миллион человеко-лет, а при МБК этот показатель составляет 5 случаев на млн человек. В Японии УБК диагностируется реже – в 30% случаев, а МБК – в 70%, причина такого расхождения остается неясной. В описанной когорте были зарегистрированы несколько пациентов с ВИЧ, в связи с чем высказывается предположение, что региональные различия в причинах MБК, возможно, связаны с низкой заболеваемостью НHV8 в Японии по сравнению с другими странами [14, 15]. У ВИЧ-негативных пациентов частота НHV8-негативного варианта МКБ составляет 2–50% случаев и, вероятно, определяется распространенностью НHV8 в отдельном регионе или стране. Почти у всех ВИЧ-позитивных пациентов МКБ ассоциирована с НHV8 [5].
БК встречается в любом возрасте (диапазон – 2–84 года), при этом начало заболевания УБК чаще приходится на четвертое десятилетие (средний возраст – 34 года), а средний возраст начала заболевания МБК – от пятого до седьмого десятилетия без каких-либо гендерных различий [16, 17]. Ожидаемая продолжительность жизни, как правило, не изменяется после установления диагноза УБК. Тем не менее пациенты могут подвергаться повышенному риску развития паранеопластической пузырчатки и лимфом. При МБК характер клинических проявлений и течение заболевания в значительной степени зависят от эндемичности НHV8 в общей популяции [15, 17–19]. По данным 20-летнего исследования во Франции 273 пациентов с гистопатологией л/у, соответствующей БК, 18 пациентов были исключены из исследования в связи с диагностированными злокачественными гематологическими заболеваниями. У пациентов с УБК 2-летняя выживаемость составила 98,1%, а при иМБК – 100% [20].
Клиническая картина БК
Клиническая картина заболевания часто может помочь в сложной диагностике различных подмножеств БК.
Уницентрический (одноцентровый) вариант. В большинстве случаев УБК протекает бессимптомно, поскольку опухолевидное образование растет медленно – годами, достигая значительных размеров; диагноз часто является случайным. У некоторых (16%) пациентов наблюдаются незначительно выраженные воспалительные симптомы, такие как усталость и потеря массы тела. Как правило, симптомы, обусловленные сдавливанием жизненно важных структур (например, дыхательных путей, сосудисто-нервных пучков или мочеточников), возникают в развернутой стадии заболевания. В большой серии случаев УБК наиболее частыми областями поражения являются средостение (29%), шея (23%), брюшная/тазовая полость (21%) и забрюшинное пространство (17%). Лабораторные анализы обычно в пределах референсных значений, но иногда диагностируют анемию, гипергаммаглобулинемию и ускорение СОЭ. При морфологическом исследовании л/у в 70–90% случаев выявляют гиалиново-васкулярный подтип гистопатологии [5, 16, 20–22].
Основным критерием УБК является поражение одного или цепочки л/у в пределах одной анатомической зоны. Следовательно, диагностическое исследование должно включать не только гистологическое исследование удаленного л/у, но и рентгенологическое исследование, чаще всего с помощью компьютерной (КТ) или позитронно-эмиссионной и компьютерной томографии (ПЭТ/КТ). Гепатомегалия или спленомегалия встречается редко, и их наличие должно побудить к рассмотрению альтернативных или дополнительных диагнозов. Аналогичным образом лабораторные отклонения, такие как анемия или признаки воспаления (повышенный уровень С-реактивного белка [СРБ] или увеличение СОЭ), могут присутствовать, но встречаются редко [5].
Мультицентрический (генерализованный) вариант. Термин «мультицентрическая болезнь Кастлемана» охватывает спектр состояний, которые характеризуются многоцентровой лимфаденопатией, лабораторными отклонениями и системными воспалительными симптомами, связанными с полиорганной дисфункцией органов, часто тяжелыми или угрожающими жизни. Эти симптомы обычно быстро прогрессируют, хотя из-за редкости заболевания клиническое распознавание иногда задерживается. В то время как бессимптомный УБК встречается часто, MБК, как правило, сопровождается клиническими проявлениями [5, 23].
Основной симптомокомплекс MБК включает лихорадку, потерю массы тела, ночную потливость, недомогание, общую слабость и генерализованную лимфаденопатию с спленомегалией, которая может быть массивной, и в некоторых случаях гепатомегалию. Обычно лимфаденопатия генерализована, но имеет относительно небольшой объем, объемная аденопатия встречается редко. Отеки, достигающие степени анасарки (плевральный выпот и асцит при КТ), являются результатом гипоальбуминемии и, возможно, вызванной цитокинами сосудистой дисрегуляции, могут приводить к внутрисосудистому истощению и нарушению функции почек даже при наличии значительной внесосудистой перегрузки жидкостью.
Мультицентрическая КБ характеризуется широким спектром изменений лабораторных показателей, таких как анемия, тромбоцитопения/тромбоцитоз, лейкоцитоз, ускорение СОЭ, увеличение уровней СРБ, циркулирующих иммунных комплексов, поликлональная гипергаммаглобулинемия, протеинурия. В течение симптоматических периодов повышается уровень маркеров воспаления. Воспалительные симптомы могут быть эпизодическими, проявляются «вспышками», причины которых четко не установлены. В более тяжелых случаях, как только воспалительная вспышка установлена, она становится самоподдерживающейся и может быть опасной для жизни [5, 18, 23–26].
Прочие особенности МБК могут включать аутоиммунные, гемофагоцитарные, воспалительные или идиопатические причины цитопений, гепатоспленомегалию, периферическую невропатию; легочную патологию, такую как появление инфильтратов, рестриктивные заболевания легких, лимфоидный интерстициальный пневмонит и облитерирующий бронхиолит, а также появление кожных высыпаний, гиперпигментации, гемангиоматоза, паранеопластической пузырчатки и саркомы Капоши. Тяжесть симптоматики бывает разной и может со временем меняться [10, 11, 18, 23, 24, 27]. Экстранодальные поражения (псевдоопухолевые образования) редки и встречаются в области орбит, носоглотки, тонкой кишки, подкожной клетчатки и скелетных мышцах, глубоких мягких тканях грудной клетки, шеи и в ретроперитонеальной области [23, 27–33].
Мультицицентрическая БК включает клинический подтип, связанный с HHV8, и идиопатическую форму, негативную по отношению к HHV-8. Впервые HHV8 был обнаружен у группы пациентов, имевших сопутствовавший POEMS-синдром, который в классическом варианте представляет собой редкий вариант паранеопластического синдрома, чаще всего связанного с остеосклеротической миеломой. Несколько определяющих признаков содержатся в аббревиатуре POEMS – периферическая невропатия, органомегалия (гепатоспленомегалия), эндокринопатия, моноклональная гаммапатия (обычно λ-легкая цепь) и изменения кожи. Другие важные особенности, не включенные в аббревиатуру, проявляются склеротическими поражениями костей, внесосудистой перегрузкой объемом, тромбоцитозом, лимфаденопатией и аномальными тестами функции легких [34].
Как правило, HHV8-положительная БК обнаруживается у людей, живущих с ВИЧ-инфекцией (которая может хорошо контролироваться подавленной вирусной нагрузкой ВИЧ и адекватным восстановлением CD4+-Т-клеток), но может быть обнаружена и в отсутствие ВИЧ-инфекции. В единичных случаях HHV8-отрицательная МКБ диагностируется у иммунокомпрометированных лиц и может быть обнаружена в сочетании с другими новообразованиями, включая саркому Капоши, В-клеточные лимфомы и лимфому Ходжкина [34–36].
Идиопатическая МБК (иМБК) отличается прогрессирующим течением, особенно агрессивным при наличии синдрома TAFRO, который иногда считается отдельным заболеванием, первоначально описанным в Японии и позже зарегистрированным по всему миру [37]. Основываясь на классификации тяжести БК, тяжелые случаи иМБК часто сопровождаются подтипом иМБК-TAFRO с более высоким уровнем смертности, особенно в течение первых нескольких месяцев заболевания. У этих пациентов наблюдаются тромбоцитопения, асцит, лихорадка, ретикулиновый фиброз, органомегалия и, как правило, нормальный уровень иммуноглобулинов, а также смешанные или гиалиновые сосудистые гистопатологические подтипы в л/у [26, 38–41].
Существует множество предположений о том, является ли MБК аутоиммунным, инфекционным, реактивным или клональным заболеванием. Несмотря на отсутствие единого мнения, заслуживает внимания предложенная новая концептуальная модель патогенеза МБК, согласно которой как локальные, так и системные проявления заболевания обусловлены нарушением регуляции активности цитокинов и представляют собой влияние реактивных изменений повышенных уровней интерлейкина-6 (ИЛ-6) и в меньшей степени других циркулирующих факторов цитокинового и хемокинового шторма. Несмотря на то что основные механизмы, ответственные за цитокиновый шторм в случаях иМКД, остаются гипотетическими, важная патогенная роль HHV8 (Human Herpes Virus 8) неоспорима в случаях иМКД, ассоциированных с HHV-8. У этих пациентов иммуносупрессия, обусловленная HHV8, нарушает иммунный контроль хозяина. Повышенная литическая репликация запускает каскады противовирусной сигнализации, которые приводят к избыточной продукции человеческого ИЛ-6 (чИЛ-6) и других цитокинов, включая ИЛ-10, фактор некроза опухоли-α (ФНО-α) и ИЛ-1 [24].
При MКБ, ассоциированной с HНV8 (примерно 50% случаев), гиперактивность цитокинов вызывается вирусами, что также может приводить к атипичной пролиферации л/у и потенциальной их трансформации в лимфому. Гиперцитокинемия может быть результатом различных патологических процессов, которые в конечном итоге приводят к различным сочетаниям клинических проявлений и разнообразной патологии в лимфоидных тканях [25].
Поражение почек при БК
Поражение почек при БК встречается редко, клиническими проявлениями которых могут быть мочевой и нефротический синдромы, а также прогрессирующая почечная недостаточность (ПН). Так, X.G. Yuan et al. [42] отмечали симптомы поражения почек в 12% наблюдений до установления БК, синхронно (48%) или позже (40%). Наиболее распространенными проявлениями поражения почек были нефротический синдром (61%, n=46) и хроническая ПН (45%, n=34).
Гломерулярные заболевания почек с морфологической картиной мембранопролиферативного гломерулонефрита (МПГН) наиболее характерны для синдромов TAFRO и POEMS и связаны с гиперпродукцией цитокинов и факторов роста, таких как ИЛ-6 и VEGF (фактор роста эндотелия сосудов) [43–46]. При этом варианте поражения морфологические признаки МПГН, такие как дольчатость клубочков, отек эндотелиальных клеток, гиперклеточность мезангия и удвоение контуров гломерулярной базальной мембраны, не сопровождаются экспрессией иммуноглобулинов и компонентов системы комплемента, характерных для иммунокомплексного гломерулонефрита либо комплемент-ассоциированного МПГН [47–50]. Большинство авторов рассматривают подобные изменения как проявление хронической тромботической микроангиопатии (ТМА) аналогично развитию ТМА-подобных изменений при преэклампсии и у пациентов, получающих лечение анти-VEGF-препаратами. Характерной морфологической особенностью этих изменений является диффузный отек эндотелия (эндотелиоз) и мезангиолиз. Основным патогенетическим механизмом принято считать подавление продукции гломерулярного VEGF подоцитами и связанное с этим повреждение эндотелия [51].
Тубулоинтерстициальные нефриты в виде изолированных неспецифических воспалительных инфильтратов стромы и канальцев встречаются редко, чаще описываются при плазматическом варианте генерализованной формы БК и в сочетании с поражением клубочков [52].
Гидронефроз вследствие сдавления мочеточников л/у брюшной полости чаще всего встречается при гиалиново-васкулярном варианте УБК [53–55]. Опухолевидные инфильтраты почек описаны как при гиалиново-васкулярном, так и при плазмоклеточном варианте, и в таких случаях проводится дифференциальный диагноз со злокачественными новообразованиями [56–59].
Амилоидоз. Развитие амилоидоза характеризуется отложением нерастворимых амилоидных фибрилл в различных тканях. Тип и частота поражения органов варьируются в зависимости от подтипа амилоидоза, определяемого белком-предшественником. Сывороточный амилоидный А-протеин (SAA) является предшественником белка при амилоидозе АА (АА) и его продукция индуцируется медиаторами воспаления, такими как ФНО-α, ИЛ-1 и ИЛ-6, которые хронически и устойчиво повышены при широком спектре хронических воспалительных заболеваний, включая БК. Хотя этиология иМБК неизвестна, гиперпродукция ИЛ-6 индуцирует преимущественно печеночную продукцию реагентов острой фазы, включая SAA, может приводить к увеличению транскрипции SAA и развитию амилоидоза АА как редкого осложнения [60].
Что касается УБК, причина АА-амилоидоза (AA-А) менее ясна, поскольку при этом заболевании отмечают небольшое увеличение или отсутствие воспалительных цитокинов. Однако A. Fayand et al. [61] показали, что АА-А наиболее часто осложняет УБК у пациентов с выраженными конституциональными симптомами и воспалительным синдромом. Это согласуется с наблюдаемым преобладанием плазмоклеточного или смешанного гистологического подтипа, которые обычно характеризуют иMБК. Таким образом, авторы предположили, что УБК, характеризующаяся плазмоклеточной гистологией и избыточной продукцией ИЛ-6, возможно, более близкими к иMБК, чем к классическому варианту УБК, могут вызывать AA-A.
При иМБК и УБК, ассоциированных с развитием АА-А, характерен преимущественно плазматический вариант гистологического строения л/у [61–63]. Было высказано предположение о наличии у некоторых пациентов специфического аутовоспалительного состояния, приводящего к иМБК и AA-А. Предложенная гипотеза состоит в том, что АА-А может возникать у людей с генетической предрасположенностью, такой как полиморфизм SAA1, ранее описанный у пациентов с ревматоидным артритом [64, 65].
L. Bernabei et al. проанализировали опубликованные в литературе случаи УБК (n=39) и МБК (n=22), связанные с АА-А [60]. Авторы отметили, что АА-А был установлен одновременно с диагнозом УБК у 72% пациентов, до установления диагноза УБК – в 10% наблюдений и после первоначальной диагностики УБК – у 13% пациентов. Средний возраст пациентов составил 42 года, при этом 59% составляли женщины. Гистопатологический подтип изменений в л/у был преимущественно плазмоцитарным (67%), смешанным и гиалиновым сосудистым (23 и 8% соответственно). Наиболее часто пораженными амилоидозом органами были почки (59%) и печень (49%), желудочно-кишечный тракт (21%), л/у (15%) и селезенка (15%). Наличие амилоида более чем в одном органе было выявлено в половине всех наблюдений. Имеются лишь единичные описания клинических наблюдений AL-амилоидоза у пациентов с плазмоклеточным типом БК, AH-амилоидоза при лимфоплазмоцитарной лимфоме и АА-А при гипер-IgD синдроме [66, 67].
Мы представляем клиническое наблюдение, иллюстрирующее особенности течения и трудности диагностики и лечения БК, осложненного развитием АА-А.
Клинический случай
Пациентка Ш. 29 лет поступила в нефрологическое отделение ГКБ № 52 ДЗМ в ноябре 2017 г. для верификации характера почечного поражения. Из анамнеза известно, что с 12-летнего возраста больную беспокоили боли в правой половине живота, поясничной области, повышение температуры до 40◦С, которые трактовались как рецидивирующий хронический пиелонефрит с частыми обострениями не менее 2–3 раз в год. В возрасте 19 лет при амбулаторном обследовании была выявлена железодефицитная анемия, ускорение СОЭ. При уьтразвуковом исследовании (УЗИ) диагностирована внутрибрюшная лимфоаденопатия без последующего уточнения диагноза. Медицинская документация отсутствует.
В возрасте 22 лет первая беременность осложнилась отслойкой плаценты на 41-й неделе гестации, в 25-летнем возрасте при второй беременности в анализах мочи выявлена протеинурия (уровень неизвестен) и проведено родоразрешение на сроке гестации 36–38 недель путем кесарева сечения. Через год после вторых родов (июнь 2016) в связи с сохраняющейся протеинурией от 0,3 до 1,2 г/л без изменений в мочевом осадке была госпитализирована в нефрологическое отделение по месту жительства. В анализах крови: гемоглобин (Нb) – 74 г/л, тромбоциты – 539 тыс/мкл, СОЭ – 7 мм/ч, повышение уровня мочевины (14,7 ммоль/л) и креатинина сыворотки (215 мкмоль/л). Скорость кубочковой фиьтрации – 26 мл/мин (по CRD-EPI). При КТ органов брюшной полости и почек выявлена двусторонняя каликопиелоэктазия, асимметрия размеров почек (правая почка – 105х53 мм, левая – 125×53 мм). Выставлен диагноз «хронический гломерулонефрит, хроническая ПН» . Проводилась терапия эритропоэз-стимулирующими препаратами. В связи с прогрессированием азотемии, нарастанием уровня креатинина до 480 мкмоль/л 31.10.2016 начато лечение программным гемодиализом.
При поступлении состояние средней степени тяжести, стабильное, больная нормального питания. Сознание ясное. В правой подключичной области установлен туннельный центральный венозный катетер, выходное отверстие без признаков воспаления. Кожные покровы и видимые слизистые оболочки бледные, отеков нет. Периферические л/у не пальпируются. В легких везикулярное дыхание, проводится во все отделы, побочные дыхательные шумы не выслушиваются. Сердечные тоны ясные, ритм правильный, систолический шум в точке Боткина. Частота сердечных сокращений – 76 в 1 минуту. Артериальное давление – 130/80 мм рт.ст. Язык влажный, обложен у корня сероватым налетом. Живот не увеличен в объеме, участвует в акте дыхания, при пальпации мягкий, безболезненный во всех отделах, перистальтика выслушивается. Печень по краю реберной дуги, селезенка не пальпируется. Диурез – 1000 мл, дизурии нет, стул регулярный.
В общем анализе крови сохраняется анемия: эритроциты – 3,0×1012/л, Нb – 79 г/л, лейкоцитоз до 11,0х109/л, тромбоцитоз до 465×109/л, СОЭ – 50 мм/ч. Мочевина – 33,8 ммоль/л, креатинин сыворотки – 615,9 мкмоль/л, СРБ – 41,5 мг/л, ферритин – 630 мкг/л, общий белок – 78 г/л, альбумин – 36 г/л, калий сыворотки – 5,9 ммоль/л, мочевая кислота – 508 мкмоль/л. В общем анализе мочи: белок – 1,7 г/л, эритроциты – 60 кл/мкл, лейкоциты – 11 кл/мкл, суточная протеинурия – 4,05 г.
В связи с эпизодами немотивированного субфебрилитета, анемии, сохраняющейся на протяжении нескольких лет до развития ПН, наличием лейкоцитоза, тромбоцитоза, ускорения СОЭ, увеличения уровней СРБ, ферритина, титра антинуклеарного фактора (АНФ) – 1/160, протеинурии нефротического уровня и быстропрогрессирующей ПН (в течение нескольких месяцев), потребовавшей проведения диализа, проводился дифференциальный диагноз с целью исключения системных заболеваний.
При обследовании: АНФ – 1/160, тип свечения – крапчатый. Иммуноглобуин G (IgG) – 1505 мг/дл, IgA – 560 мг/дл.
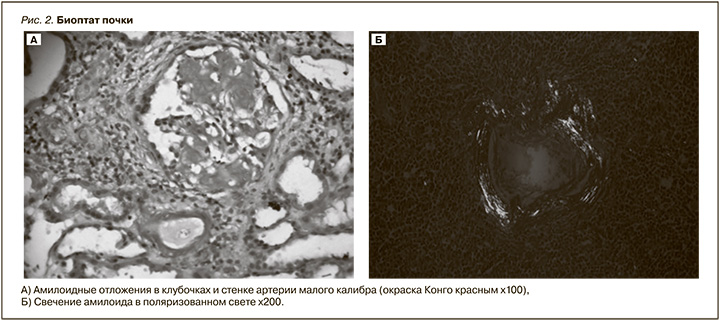
Выполнена пункционная биопсия почки (рис. 2). В препарате 14 клубочков, 5 из которых полностью склерозированы. Оставшиеся клубочки обычных размеров, в большинстве из них определяются массивные отложения эозинофильных, PAS-негативных бесклеточных масс в мезангии и в стенке капиллярных петель. Диффузный фиброз интерстиция и атрофия канальцев, занимающие более 70% площади паренхимы. Сохранные канальцы гипертрофированы. Определяются массивные отложения эозинофильных, PAS-негативных бесклеточных масс в интерстиции. Неспецифическая лимфогистиоцитарная инфильтрация интерстиция в зонах склероза. B артериях и артериолах отмечается циркулярное утолщение стенок сосудов за счет отложения эозинофильных бесклеточных масс. Окраска Конго красным – позитивное окрашивание материала, инфильтрирующего клубочки, сосуды. При исследовании в поляризованном свете отмечается яблочно-зеленое свечение в проекции отложения бесклеточных масс. При иммунофлюоресцентном исследовании IgG, IgM, IgA C3, C1q, Kappa, Lambda – негативно. По данным морфологического исследования был диагностирован АА-А.
При КТ-исследовании органов брюшной полости (рис. 3) кпереди от ворот селезенки определяется образование овоидной формы с ровными четкими контурами, размером 4,5×4,0×3,0 см, менее активно, чем селезенка, накапливающее контрастное вещество. Рядом – группа умеренно увеличенных л/у с максимальными аксиллярными размерами 23×13 мм. Определяется цепочка умеренно увеличенных парааортальных забрюшинных л/у, наибольший – на уровне ворот левой почки – 20,7×9,7 мм. Правая почка умеренно уменьшена в размерах (9,5×3,0×5,0 см), в паренхиме – мелкие одиночные кисты до 5 мм. Под нижним полюсом левой почки – гематома малого объема (до 5 мл), частично накапливающая контрастное вещество. Селезенка обычной формы и размера (вертикальный размер –8,7 см) с четкими ровными контурами, структура и плотность не изменены. Печень увеличена с ровными контурами, плотность паренхимы – 54,5 HU, паренхима накапливает контрастное вещество равномерно, зоны патологического накопления контрастного вещества не определяются.
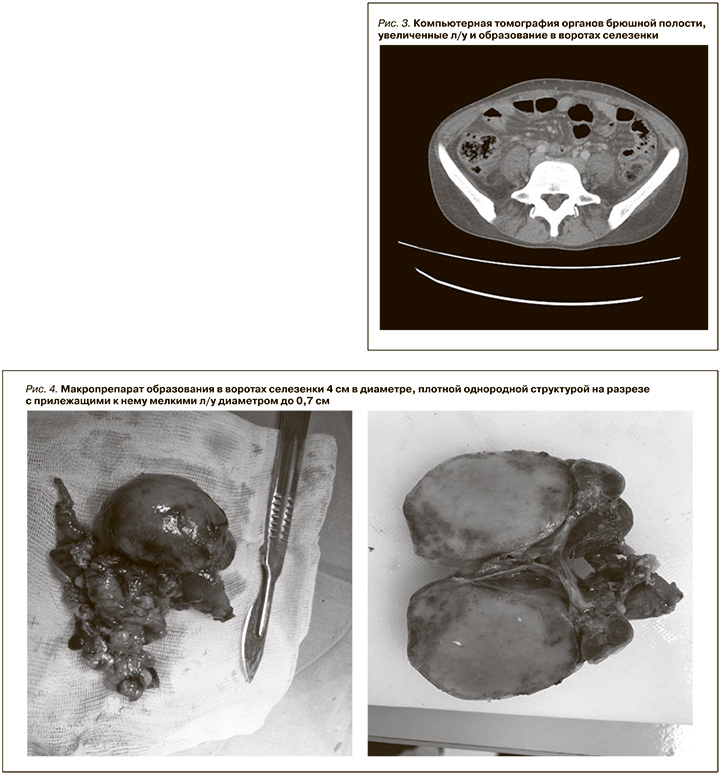
Заключение: увеличенные л/у в воротах селезенки, образование в воротах селезенки (увеличенный лимфоузел? добавочная долька селезенки?). Умеренно выраженная ретроперитонеальная лимфаденопатия. Гепатомегалия. Гематома малого объема под нижним полюсом левой почки (после биопсии почки).
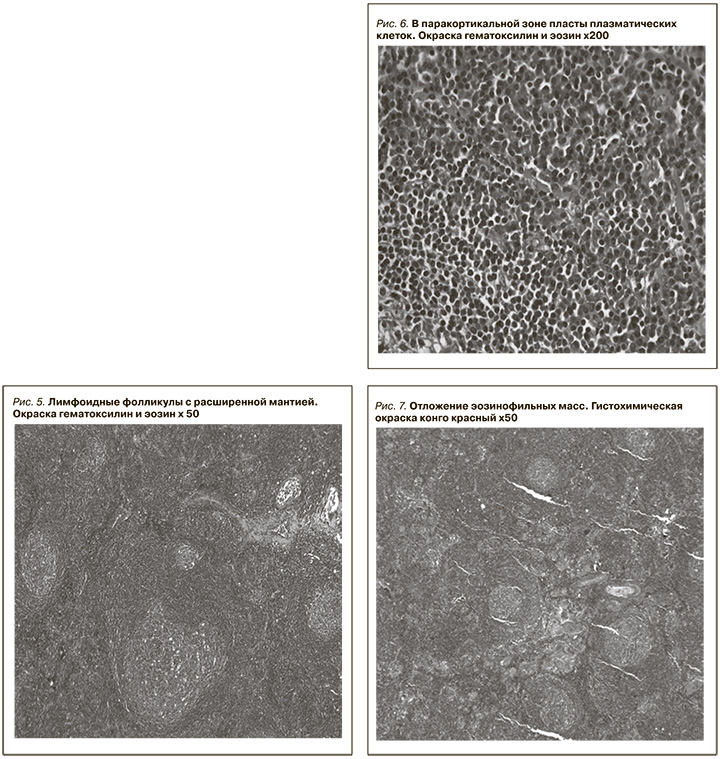
С учетом наличия лимфоаденопатии, воспалительного синдрома, данных гистологического исследования почечного биоптата заподозрена БК, в связи с чем через лапароскопический доступ удалено образование в области ворот селезенки, макроскопически выглядящее как добавочная долька селезенки (рис. 4).
В лабораторию доставлено новообразование округлой формы диаметром 4 см, темно-коричневого цвета, на разрезе к нему прилежали мелкие л/у диаметром до 0,7 см. Гистологически оба препарата имели строение л/у с измененными фолликулами, гиалинозом и отложением оптически плотных масс (амилоид?). Паренхима была представлена гиперплазированной лимфоидной тканью с примесью плазматических клеток. Была заподозрена БК, смешанный вариант.
Для уточнения диагноза материал был консультирован в патологоанатомическом отделении ФГБУ «НМИЦ гематологии» МЗ РФ. В гистологических препаратах, изготовленных из всех представленных блоков: в срезах ткани л/у гистоархитектоника нарушена, фолликулы с регрессивно трасформированными или широкими светлыми центрами размножения, окруженные преимущественно расширенной зоной мантии (рис. 5). Интерфолликулярно в паракортикальной зоне определяются пласты зрелых плазматических клеток, краевой и промежуточные синусы расширены с наличием мелких лимфоидных элементов, плазмоцитов (рис. 6). В стенках мелких сосудов определяются отложения эозинофильных масс, которые при окраске Конго красным и исследовании в поляризованном свете имеют яблочно-зеленое свечение (рис. 7 и 8). На срезах с парафинового блока проведено иммуногистохимическое исследование (2632/17) с использованием антител к CD-3, CD-20, IgG, IgG4, EBER (ISH), HHV-8 (LANA-1), IgL. Фолликулы представлены В-зоной CD20+, EBER-позитивных клеток или позитивных HHV8 не обнаружено. Интерфоликулярно присутствуют пласты зрелых плазматических клеток. IgG+, Kappa+, Lambda+, соотношение IgG4/IgG <30% (рис. 9–12). Морфологическая картина и иммунофенотип характеризуют субстрат БК, плазмоклеточный вариант, HHV8 с вторичным амилоидозом.
Выставлен диагноз «идиопатическая мультицентрическая HHV8-негативная БК, плазмоклеточный вариант. АА-амилоидоз с поражением почек, л/у. Хроническая ПН, программный гемодиализ с октября 2016 г.».
С учетом полученных данных проведена терапия ритуксимабом (500 мг однократно), рассматривался вопрос о введении тоцилизумаба по месту жительства. При динамическом обследовании через 2 года при КТ органов грудной клетки, брюшной полости лимофаденопатия не выявлена. По данным эхокардиографии (Эхо-КГ) сократительная функция миокарда составляла 46%, выявлен диффузный гипокинез. Лабораторно отмечалась положительная динамика в виде снижения уровня СРБ 1,9 мг/л (норма до 6 мг/л), уровень Нb – 117 г/л, нормализация уровня тромбоцитов – 201×109/л, нормализация СОЭ – 11 мм/ч, ферритина – 267 мкг/л. Суточная протеинурия – 1,2 г, ИЛ-6 – 5,95 пг/мл (норма), IgG – 1121 мг/дл, Ig А – 423 мг/дл. При полимеразной цепной реакции вирус герпеса 8-го типа не обнаружен. Сохранялась диализ-потребная ПН. Констатировано, что на фоне хирургического лечения и применения ритуксимаба отмечалось уменьшение активности воспаления: снижение СОЭ, лейкоцитоза, уровней СРБ, ферритина.
В июне 2022 г. пациентка госпитализирована в отделение нефрологии с целью динамического контроля. При УЗИ органов брюшной полости признаки умеренных диффузных изменений поджелудочной железы, свободная жидкость в малом тазу в небольшом количестве, утолщение стенок нисходящего отдела ободочной кишки. Размеры почек: правой – 78×36 мм, левой – 83х41 мм, паренхима справа – 8 мм, слева – 10 мм, кисты почек. Выполнена мультиспиральная компьютерная томография (МСКТ) брюшной полости и забрюшинного пространства с контрастным усилением: признаки гепатомегалии, застойных изменений в печени, простой кисты в правой почке, сморщенных обеих почек, удвоение правой ЧЛМС?, кистозное образование в левом яичнике, следовой выпот в малом тазу. По данным МСКТ, паращитовидных желез с внутривенным контрастированием аденомы не выявлено. По данным ультразвуковой допплерографии вен нижних конечностей, признаков тромбоза не выявлено. Лабораторно: Нв – 101 г/л, тромбоциты – 182×109/л, лейкоцитоза нет, СРБ и ИЛ-6 отрицательные, мочевая кислота – 590 мкмоль/л, нарушение кальциево-фосфорного обмена (кальций – 1,95 ммоль/л, фосфор – 2,38 ммоль/л), коагулограмма без особенностей.
По данным Эхо-КГ, снижение общей систолической функции миокарда левого желудочка (фракция выброса левого желудочка – 40%) за счет диффузного гипокинеза, выраженная симметричная гипертрофия миокарда левого желудочка, признаков легочной гипертензии не выявлено.
По заключению кардиолога выявлены клинико-инструментальные признаки вторичной рестриктивной кардиомиопатии со снижением систолической функции левого желудочка в рамках развития АА-А. С учетом основного заболевания и повышения эффективности заместительной почечной терапии предложено расширить диализную программу до трех сессий в неделю (пациентка по месту жительства получала лечение программным гемодиализом 2 раза в неделю). Была выписана.
Обсуждение
Несмотря на углубленное изучение клинико-патологических особенностей БК и лежащей в их основе дисрегуляции активности цитокинов, диагностика и точная классификация МКБ остается сложной задачей. Клиническая картина крайне неоднородна, и патологические находки не являются конкретными. Во-вторых, несмотря на то что у большинства пациентов БК представляет собой доброкачественное заболевание, пациенты подвергаются высокому риску развития определенных типов злокачественных новообразований, что может сильно затруднить проведение дифференциальной диагностики, лечения и ухудшить прогноз.
В представленном нами наблюдении клиническая картина заболевания характеризовалась длительным бессимптомным течением и дебютировало в 12-летнем возрасте рецидивирующим пиелонефритом, обусловленным нарушением пассажа мочи вследствие сдавления мочеточника увеличенными л/у. К сожалению, причина внутрибрюшной лимфоаденопатии с относительно небольшим объемом, обнаруженной при УЗИ, не была выяснена. Заболевание первоначально характеризовалось неагрессивным течением с длительными периодами стабилизации состояния, слабостью и появлением протеинурии во время беременности. В 26-летнем возрасте через год после вторых родов на фоне сохраняющейся протеинурии в диапазоне от 0,3 до 1,2 г/л, появились конституциальные симптомы заболевания в виде слабости, немотивированного субфебрилитета. Впервые появился широкий спектр изменений лабораторных показателей таких как анемия, тромбоцитоз, ускорение СОЭ, увеличение уровней СРБ, ферритина, поликлональная гипергаммаглобулинемия, увеличение протеинурии до нефротического уровня со скудным мочевым осадком, повышение уровня азотистых шлаков с быстрым прогрессированием нарушения функции почек до терминальной ПН, потребовавшей заместительной почечной терапии диализом. Протеинурия, воспалительный синдром, наличие лимфоаденопатии и развитие органной недостаточности потребовали незамедлительного рассмотрения вопроса о наличии БК, ассоциированной с развитием АА-А. Предположение было подтверждено гистологическим исследованием л/у и почечного биоптата.
Несмотря на развитие амилоидоза почек и терминальной ПН, с учетом наличия воспалительного синдрома предпринято хирургическое удаление гиперпластических л/у и терапия ритуксимабом с целью достижения клинической и биологической ремиссии заболевания.
В течение длительного времени основными методами лечения БК считались радикальное хирургическое удаление увеличенных л/у и проведение цитостатической терапии в соответствии с протоколом лечения лимфопролиферативных заболеваний. Наиболее часто применялись курсы полихимио-терапии, используемые при неходжкинской лимфоме, в т.ч. применение моноклонального химерного антитела – ритуксимаба. Описаны успешные случаи предоперационной лучевой терапии с целью уменьшения размеров особо крупных л/у в сочетании с глюкокортикостероидами.
По мере изучения патогенеза заболевания стала очевидной роль гиперпродукции цитокинов (прежде всего ИЛ-6), а также вирусных агентов в развитии БК, что позволило использовать препараты направленного действия. По современным представлениям [8], пациентам с БК рекомендуется лечение противовирусными препаратами (валганцикловир, зидовудин), антителами к ИЛ-6 (силтуксимаб) и к рецептору ИЛ-6 (тоцилизумаб), моноклональными антителами к CD20 (ритуксимаб). Отмечено, что монотерапия ритуксимабом может усугубить или выявить саркому Капоши, и именно поэтому к ритуксимабу добавляют пегилированный липосомальный доксорубицин при наличии сопутствующей саркомы Капоши. Однако, несмотря на то что моноклональные антитела, направленные на ИЛ-6, были эффективными для некоторых пациентов с иМБК, большинство из которых не достигали длительной ремиссии, в связи с чем требуются новые методы лечения.
Заключение
Имеющиеся в литературе сведения до конца не раскрывают сути проблемы, касающейся диагностики, выбора лечебной тактики и дифференциально-диагностических аспектов БК. Необходимы дальнейшие хорошо спланированные проспективные исследования в рамках углубления знаний о патофизио-логии БК с целью разработки эффективных методов лечения пациентов с разными вариантами заболевания.


