
Гемолитико-уремический синдром (ГУС) служит наиболее частой причиной острой почечной недостаточности (острого повреждения почек по новой терминологии) у детей. Он характеризуется триадой признаков: микроангиопатической (Кумбс-негативной) гемолитической анемией с наличием фрагментированных эритроцитов (шизоцитов), тромбоцитопенией и острой почечной недостаточностью [1–3].
В основе ГУС лежит тромботическая микроангиопатия (ТМА) – своеобразный синдром, основные формы которого различаются патогенетическими механизмами, но имеют общие клинические проявления и гистологическую картину. Независимо от механизмов развития симптомокомплекс ГУС является следствием поражения эндотелиальных клеток с последующей утратой ими тромборезистентности, активацией тромбоцитов с образованием тромбов в микроциркуляторном русле, преимущественно почек, механическим повреждением эритроцитов в результате контакта с многочисленными тромбами [2, 4].
У большинства (90–95%) детей отмечается т.н. типичный, или, как его ранее называли, постдиарейный ГУС (Д+ГУС), вторичный по отношению к инфекции Escherichia coli, продуцирующей шигатоксин (Shigatoxine; Stx) (Stx – продуцирующая E. coli, STEC-HUS). Поскольку диарея нередко предшествует и другой форме ГУС, называемой атипичной (аГУС, Non-STEC-HUS), сегодня термин «постдиарейный ГУС (Д+ГУС)» следует считать устаревшим. Атипичный ГУС встречается гораздо реже и служит результатом генетической аномалии белков, регулирующих процесс активации альтернативного пути комплемента или реже – выработки антител к фактору Н – основному регуляторному протеину этого пути [1, 2, 5].
Атипичный ГУС составляет 5–10% от всех случаев ГУС у детей, является прогрессирующим генетическим заболеванием с крайне высоким риском внезапной смерти и необратимых инвалидизирующих повреждений жизненно важных органов, включая не только почки, но и головной мозг, сердце и печень. Поскольку в основе аГУС лежит ТМА, связанная с неконтролируемой активацией системы комплемента, синонимом термина аГУС является термин «комплемент-зависимая ТМА». Основным органом-мишенью микроангиопатического тромбообразования при аГУС, как и при типичном ГУС, служат почки, однако не менее чем в 20% случаев происходит генерализация ТМА, приводящая к развитию ишемического повреждения органов с картиной полиорганной недостаточности [2, 6–8], что позволяет считать аГУС системным заболеванием.
АГУС – хроническое заболевание, требующее пожизненного наблюдения и лечения. Заболеваемость и распространенность аГУС изучены не до конца. По данным различных источников, в странах с достаточным уровнем диагностики распространенность заболевания составляет от 2 до 7 пациентов на 1 млн населения (детей и взрослых) [2–4].
Начало заболевания, как правило, внезапное. К первым проявлениям у детей раннего возраста могут относиться бледность, общее недомогание, плохой аппетит, рвота, сонливость, отеки. Экстраренальные симптомы заболевания зависят от преимущественного поражения того или иного органа. Наиболее часто поражается ЦНС (20–43% пациентов), что проявляется судорогами, сопором, комой. В патогенезе энцефалопатии помимо собственно ТМА головного мозга играют важную роль одновременно несколько факторов: отек и кровоизлияния в вещество мозга, гипоксия, артериальная гипертензия, электролитные расстройства, метаболические нарушения, системная воспалительная реакция. Массивное внутрисосудистое потребление тромбоцитов может провоцировать развитие коагулопатии потребления: гипер- или гипокоагуляцию, гипопротромбинемию, гипофибриногенемию, повышение уровня продуктов деградации фибрина, Д-димера, замедленный фибринолиз, нарушение агрегации тромбоцитов.

У большинства пациентов при первом лабораторном исследовании выявляется полная диагностическая триада ГУС: тяжелая анемия (гемоглобин 90–40 г/л), тромбоцитопения (<150 тыс/мкл крови) с отсутствием или небольшим риском геморрагических осложнений и нарушение функции почек (креатинин>110 мкмоль/л) с наличием или отсутствием анурии или олигурии. Мочевой синдром, как правило, представлен изолированной протеинурией, однако протеинурия может и отсутствовать. Наблюдается лейкоцитоз и умеренно выраженный ретикулоцитоз. Отрицательная реакция Кумбса при ГУС подтверждает неиммунный характер анемии, так же как и наличие шизоцитов в сочетании с высоким уровнем лактатдегидрогеназы (ЛДГ), подтверждает микроангиопатический внутрисосудистый генез гемолиза. В случае запоздалой диагностики могут наблюдаться опасные для жизни осложнения: гиперкалиемия (≥6 ммоль/л), ацидоз (сывороточный бикарбонат <15 ммоль/л) и перегрузка сосудистого русла объемом с развитием артериальной гипертензии (АГ) и гипонатриемии (<135 ммоль/л). Артериальная гипертензия встречается часто, имеет тяжелое течение и связана как с задержкой натрия и воды в случае олиго-/анурии, так и с вторичной по отношению к почечной ТМА гиперренинемией, обусловленной ишемическим повреждением почек. Возможно развитие сердечной недостаточности или неврологических осложнений (судороги) вследствие гипертензии. Большинство пациентов детского возраста нуждаются в проведении гемодиализа при поступлении. У ряда (около 20% детей) пациентов отмечается постепенное начало с субклинической анемией и колебаниями числа тромбоцитов в течение недель или месяцев при сохранной функции почек на момент постановки диагноза. У них может возникнуть спонтанная ремиссия, а затем – острый рецидив заболевания. Возможно также неуклонно прогрессирующее течение болезни, характеризующееся нарастающими АГ и протеинурией вплоть до развития нефротического синдрома и повышением уровня креатинина сыворотки в течение нескольких недель или месяцев. У некоторых пациентов анемия или тромбоцитопения может отсутствовать и единственными проявлениями почечной ТМА в таком случае будут служить АГ, протеинурия и прогрессирующее нарастание креатинина сыворотки. В этих случаях можно говорить о т.н. еполной ТМА. Таким образом, аГУС многолик по своим клиническим проявлениям и прогнозу [1–8].
Диагноз аГУС устанавливают на основании выявления симптомокомплекса ТМА при исключении Stx-ГУС и тромботической тромбоцитопенической пурпуры (ТТП), а у взрослых пациентов – еще и различных вторичных форм ТМА (системные заболевания, в т.ч. системная красная волчанка [СКВ] и антифосфолипидный синдром [АФС], ТМА, ассоциированные с беременностью и родами, приемом некоторых лекарств, злокачественными опухолями, ВИЧ-инфекцией).
Таким образом, у больных с признаками ТМА следует констатировать: 1) отсутствие критериев ГУС, связанного с Шига-токсином (посев кала и ПЦР на Шига-токсин; серология на антилипополисахаридные антитела); 2) отсутствие критериев ТТП (активность ADAMTS13 сыворотки более 5%). Примерно у трети больных подтверждением диагноза аГУС может стать снижение С3-компонента комплемента в крови. Другие изменения в системе комплемента (концентрация в плазме крови факторов, регулирующих его активность) [1–3] в нашей стране пока идентифицировать невозможно. У относительно небольшой части пациентов с аГУС, преимущественно детей (10–20%), в крови выявляются антитела к фактору Н(CFH).
До настоящего времени терапией первой линии при аГУС остается свежезамороженная плазма (СЗП). Плазмотерапию проводят в режимах плазмообмена или, что менее эффективно, инфузий до достижения ремиссии [1, 3]. О последней судят по нормализации числа тромбоцитов и прекращению микроангиопатического гемолиза (нормализация ЛДГ), при этом функция почек может оставаться сниженной. Ежедневная доза плазмы колеблется от 20 до 40 мл/кг, в ряде случаев дозу можно увеличивать до 60 мл/кг/сут.
Новым стандартом лечения пациентов с аГУС в мире является применение препарата, ингибирующего систему комплемента. В настоящее время единственным препаратом с антикомплементарным действием, зарегистрированным в России, остается экулизумаб – гуманизированные моноклональные антитела к C5-компоненту комплемента. Связываясь с ним, препарат блокирует расщепление последнего С5-конвертазой, ингибируя образование анафилотоксина С5а и терминального комплекса комплемента – комплекса мембранной атаки (МАК) С5b-С9, что приводит к блокаде провоспалительного, протромботиче-ского и литического действия комплемента [1, 9]. Поскольку у больных аГУС наблюдается постоянная неконтролируемая активация комплемента, риск развития внезапных осложнений сохраняется на протяжении всей жизни. Это обосновывает длительное, возможно пожизненное, лечение экулизумабом.
В последнее время появились работы, обосновывающие применение экулизумаба пациентами (преимущественно детьми) с аГУС, вызванным выработкой антител к CFH. Приводим собственное наблюдение больной «аутоиммунным» аГУС, особенности течения которого создали определенные трудности для диагностики и лечения.
В отделении нефрологии КБУЗ «КДКБ № 1» Владивостока с 05.01.14 по настоящее время наблюдается пациентка 7 лет (02. 2008 г.р.). Девочка от 3-й беременности, 2-х родов (брат 9 лет – здоров). Перенесенные заболевания – ветряная оспа, ангины 2 раза в год, пневмония. Привита по возрасту. Реакция Манту от 15.07.13 – папула 10 мм. Наследственность отягощена по гипертонической болезни (у отца и деда).

Анамнез заболевания: 17.12.13 была проведена вакцинация против гриппа вакциной Гриппол плюс. 25.12.13 у ребенка появились катаральные явления, затем белые налеты на миндалинах, повысилась температура тела (38–37°С). В течение 4 дней получала амоксиклав, сосудосуживающие капли в нос. 30.12.13 на коже туловища появились сыпь в виде крапивницы и отечность лица, расцененные как лекарственная аллергия. Назначены дексаметазон, тавегил, адсорбенты, купировавшие эти симптомы. Однако состояние не улучшалось, сохранялись катаральные явления в зеве, появились вялость, сонливость, иктеричность кожи, вновь крапивница, а 04.01.14 – рвота, боли в животе, макрогематурия, олигурия, АГ и гиперазотемия. Больная переведена из районной больницы, куда была госпитализирована в связи с этими симптомами, в реанимационное отделение КБУЗ «Краевая детская клиническая больница № 1» Владивостока.
Состояние при поступлении крайне тяжелое, обусловленное симптомами интоксикации, абдоминальным болевым, диспепсическим и гипертензионным синдромами, анемией, острой почечной недостаточностью. Кожные покровы бледные с иктеричностью. Отеков нет. Зев спокойный. Слизистые розовые. Дыхание, ослабленное по всем полям, хрипов и одышки нет. Тоны сердца приглушены, ритмичные, тахикардия с ЧСС 110 в минуту, АД – 128/95 – 160/100 мм рт.ст. Печень +3 см из-под края реберной дуги. Селезенка не пальпируется. Диурез 520 мл, макрогематурия. Характер стула не изменен. Физическое развитие выше среднего, гармоничное (масса тела – 23 кг, рост – 117 см). В анализах: Hb – 115 г/л, тромбоциты – 161 тыс/мкл, мочевина – 21,73 ммоль/л, креатинин – 210 мкмоль/л, ЛДГ – 1780 ЕД/л, непрямой билирубин – 47,6 мкмоль/л, АСТ – 144 ЕД/л. В общем анализе мочи: белок – 3,87 г/л, лейкоциты – 22–26 в п/зр, эритроциты – 87–92 в п/зр (рис. 1). Эритроцитарные антитела, прямая и непрямая пробы Кумбса отрицательные. Исключены инфекции и паразитарные заболевания: лептоспироз, лямблиоз, токсоплазмоз, токсокароз, аскаридоз, описторхоз, эхинококкоз, хламидиоз, уреаплазмоз, микоплазмоз, герпетические инфекции. При УЗИ почек (05.01.14) обнаружено увеличение их размеров с отсутствием кровотока в кортикальной зоне.
Характер течения заболевания и клинико-лабораторные данные позволили диагностировать ТМА. Проведенное лечение включило коррекцию анемии (трансфузии эритроцитарной массы), ежедневную плазмотерапию свежезамороженной плазмы (СЗП) (инфузии СЗП в объеме 10 мл/кг № 17), заместительную почечную терапию (ЗПТ) на гемопроцессоре Мультифильтрат в режим продленной вено-венозной гемодиафильтрации (ПВВГДФ) с дальнейшим переводом на постоянную ЗПТ в режиме непрерывного вено-венозного гемодиализа (ПВВГД), антибактериальную, дезагрегантную и антикоагулянтную терапию.
Несмотря на лечение, состояние ухудшалось: наросли признаки гипергидратации, сохранялась значительная артериальная гипертензия (АД до 152/95 мм рт.ст.), на 10-е сутки от начала терапии (с 15.01.14) развилась анурия, появилась геморрагическая сыпь. Отмечено прогрессирование анемии (Hb – 64 г/л,) и тромбоцитопении (Тр. – 79 тыс мкл), нарастание азотемии (мочевина – 43,32 ммоль/л, креатинин – 428 мкмоль/л), значительное повышение ЛДГ – до 12170 ЕД/л, усиление явлений цитолиза (АСТ – 121 ЕД/л, АЛТ – 211 ЕД/л), снижение С3-компонента комплемента – до 0,650 г/л (норма – 0,9–1,8), антитромбина III – до 76%. В то же время рецидивировали боли в животе, диарея, рвота. К 19.01.14 сформировался нефротический синдром (НС) (протеинурия – 9,9 г/л, общий белок – 55,9 г/л), который требовал проведения дифференциальной диагностики между ТМА и быстропрогрессирующим ГН. Последний невозможно было исключить из-за тромбоцитопении, препятствующей выполнению биопсии почки, поэтому назначены глюкокортикостероиды (ГКС). Оставались тяжелая артериальная гипертензия (АД до 160/110 мм рт.ст.), резистентная к 3 антигипертензивным препаратам, анурия, периодические боли в животе, разжиженный стул (рис. 1, табл. 1).
В связи с неэффективностью лечения СЗП проведен диагностический поиск с целью дифференциальной диагностики между различными ТМА (типичный и атипичный ГУС, тромботическая тромбоцитопеническая пурпура-ТТП). Исследованы активность ADAMTS13 (составившая 99%, референсные значение 93–113%) и кровь на антитела к шигатоксину 1-го и 2-го тиов (результат отрицательный), что позволило исключить ТТП и STEC-ГУС. Принимая во внимание возраст ребенка, исследованы антитела к фактору Н, которые оказались повышенными до 9000% (норма до 100%).
Таким образом, на основании данных клинического и лабораторного исследования был установлен диагноз: атипичный ГУС, обусловленный антителами к фактору Н. Острое повреждение почек по Rifle IV–V степеней тяжести. Принимая во внимание неэффективность плазмотерапии и ГКС в отношении восстановления функции почек и высокий риск развития угрожающих жизни ребенка осложнений, было решено начать лечение экулизумабом.
С 27.01.14 девочка была переведена на перитонеальный диализ (ПД).
В ожидании препарата продолжалась подобранная терапия, однако 31.01.14 вновь наступило ухудшение состояния больной, проявившееся появлением отеков, усилением выраженности абдоминального болевого синдрома, учащением стула и рвоты. В то же время впервые появились признаки поражения ЦНС – у девочки дважды отмечены клонико-тонические судороги без потери сознания. Поскольку одновременно было вновь зарегистрировано снижение Hb до 74 г/л и повышение ЛДГ до 1960 ЕД в отсутствие тромбоцитопении, состояние было расценено как рецидив ТМА, что потребовало повторного применения ПВВГДФ. Девочка опять была переведена на продленный гемодиализ. В последующие 2 недели пациентке подбирался наиболее эффективный метод ЗПТ, что достигалось чередованием разных видов ЗПТ: ПВВГДФ, ПВВГД и ПД.
С 17.02.14 (более чем через 1,5 месяца от начала болезни) после вакцинации против менингококковой, пневмококковой инфекций и инфекции, вызванной гемофильной палочкой, вакцинами «Mencevax ACWY», «Пневмо-23» и «Act-HIB» соответственно, девочке начато лечение экулизумабом. Индукционный курс препарата в дозе 600 мг в/в еженедельно в соответствии с весом ребенка (20 кг) включал 5 инфузий и был завершен 14.03.14, после чего в режиме поддерживающей терапии экулизумаб вводился 1 раз в 2 недели в дозе 300 мг (согласно инструкции препарата). По настоящее время девочка получает препарат в той же дозе, поскольку вес ее практически не изменился, достигнув лишь 18 кг.
Начало лечения сопровождалось нарастанием тяжести АГ, которая приобрела характер злокачественной и трудноуправляемой: несмотря на применение препаратов разных групп (амлодипин, конкор, моноприл), цифры АД достигали порой 190/130 мм рт.ст. Персистировали абдоминальный болевой синдром и желудочно-кишечная диспепсия, (периодическая рвота 1–2 раз в день, разжиженный стул до 5–7 раз в сутки), что рассматривали как проявления ишемического поражения кишечника, сохранялась анурия. В течение первых полутора месяцев лечения экулизумабом нормализовалось число тромбоцитов (Тр. – 371 тыс/мкл), хотя ЛДГ оставалась повышенной (432–1191 ЕД/л), анемия постепенно регрессировала (Hb – 91–124 г/л). Несмотря на это, почечная недостаточность у ребенка не разрешилась: анурия сохранялась, хотя появилось выделение минимального количества мочи (10 мл в сутки после полного отсутствия в течение более месяца), девочка оставалась диализзависимой (табл. 1).
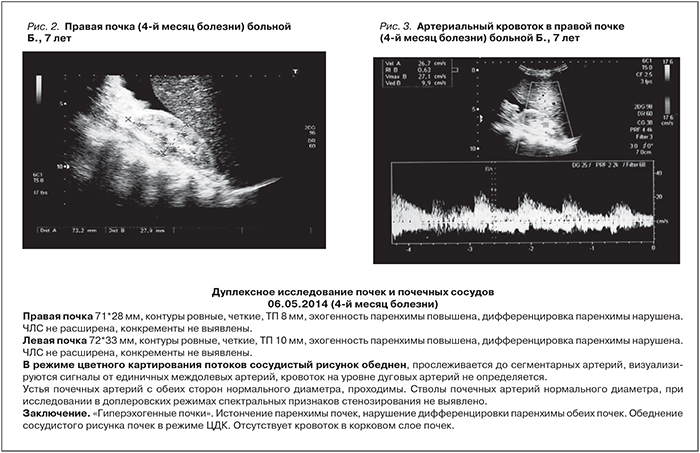
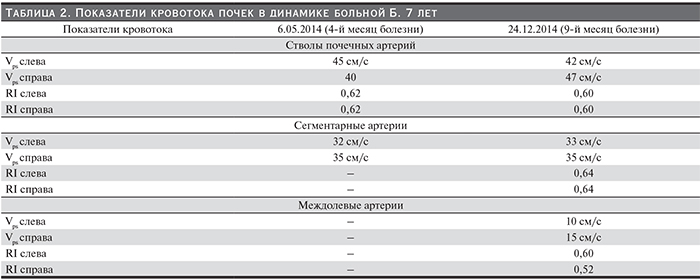
Постепенно состояние пациентки улучшилось: в мае 2014 г. (к концу 3-го месяца от начала патогенетической терапии экулизумабом) нормализовался титр антител к фактору Н (45%), стабильно нормальным стало число тромбоцитов, купировалась анемия, исчезли боли в животе, прекратились тошнота и рвота, стул стал оформленным, лучше контролировалась артериальная гипертензия (редкие подъемы АД до 160/90 мм рт.ст. при применении 4 антигипертензивных препаратов), не рецидивировал судорожный синдром. Однако почечная недостаточность не разрешалась: сохранялась анурия (диурез 30 мл/сут), мочевина – 21,14 ммоль/л, креатинин – 242 мкмоль/л. Дуплексное ультразвуковое исследование почек выявило истончение паренхимы почек, нарушение кортико-медуллярной дифференцировки, обеднение сосудистого рисунка в режиме ЦДК, отсутствие кровотока в корковом слое почек (рис 2, 3, табл. 2). На протяжении последующих 3 месяцев состояние ребенка стабилизировалось, к 8-му месяцу болезни (в августе 2014 г., спустя 6,5 месяцев применения экулизумаба) диурез увеличился до 170 мл/сут и далее стал постепенно нарастать, достигнув 200–250 мл/сут. Однако девочка по-прежнему нуждалась в диализе.
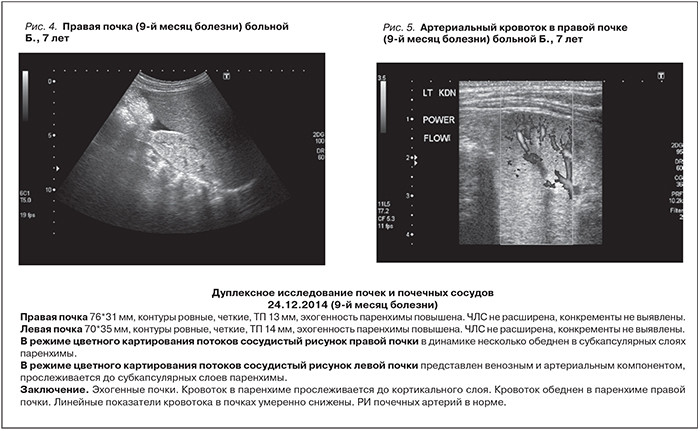
Принимая во внимание своевременное (на ранних сроках заболевания) начало лечения экулизумабом (менее 2 месяцев от начала болезни), достижение гематологической ремиссии к концу 3-го месяца терапии (Тр. 6– 583 тыс/мкл, Hb – 104–110 г/л, ЛДГ – 580 ЕД/л), нормализацию титра анти-CFH-антител (45%), непонятной оставалась причина сохраняющейся у ребенка почечной недостаточности (олигурия, креатинин крови – 242 мкмоль/л), нуждающейся в ЗПТ, в отсутствие признаков активности ТМА. В качестве объяснения сложившейся ситуации мы предположили возможность существования дополнительного фактора, который мог бы поддерживать внутриклубочковое микротромбообразование при достигнутой в ходе терапии экулизумабом блокаде комплемента. Таким фактором, с нашей точки зрения, могла быть мультигенная тромбофилия, обусловливающая склонность к развитию тромбозов любой локализации, в т.ч. и микроциркуляторных, за счет развития выраженной активации внутрисосудистого свертывания крови [10–12]. Проведенное генетическое исследование подтвердило эту гипотезу: у девочки были выявлены множественные полиморфизмы генов гемостаза (всего 6), включая гомозиготные мутации генов фибриногена (бета фибриноген [FGB]– G 455 A – G/A), ингибитора активатора плазминогена (PAI-1:4G [-675/5G] – 4G/4G) и тромбоцитарных рецепторов (Интегрин альфа-2, GPIa), а также гетерозиготные полиморфизмы всех ферментов фолатного цикла: метилентетрагидрофолатредуктазы (MTHFR) Cla429Ala (Clu/Ala); метионин-синтазы редуктазы (MTRR) – A66G (A/G); метионинсинтазы (MTR) Asp919Gly (Asp/Gly). С учетом наличия генетической тромбофилии с 15.10.14 (на 9-м месяце болезни) к лечению был добавлен фраксипарин группы низкомолекулярных гепаринов (НМГ) в дозе 0,3 мл/сут. Через неделю отмечено нарастание диуреза до 440 мл/сут. При дуплексном исследовании почек в декабре 2014 г. была выявлена значительная положительная динамика: восстановился, хотя и оставался сниженным, кровоток в корковом слое. Линейные показатели кровотока во внутрипочечных сосудах умеренно снижены (рис 4, 5, табл. 2).
В настоящее время состояние больной стабильно: гематологических признаков ТМА нет, АГ носит контролируемый характер (АД периодически повышается до 160/90 мм рт.ст., но, как правило, регистрируются нормальные его показатели), отсутствуют абдоминальный, болевой и диспепсический синдромы. Физическое развитие ребенка ниже среднего, дисгармоничное за счет низкой массы тела (масса тела – 18 кг, рост – 118 см).
Диурез составляет в среднем до 400 мл/сут, сохраняется минимальный мочевой синдром (суточная протеинурия – 0,132 г, эритроциты в пробе Нечипоренко – 4600 в 1 мл, лейкоцитов нет), СОЭ – 39 мм/ч. В биохимическом анализе крови появились изменения, характеризующие нарушения фосфорно-кальциевого обмена, закономерные для развития ХПН: паратгормон – 496,5 пг/мл (норма – 15–65), витамин Д – 18,4 нмоль/л (норма – 40–144). Девочка социально адаптирована, продолжает получать экулизумаб 300 мг в/в капельно 1 раз в 2 недели, фраксипарин 0,3 мл/сут, антигипертензивные средства (ингибиторы АПФ, бета-блокаторы, блокаторы кальциевых каналов), антианемическую терапию (рекормон, феррум лек, фолиевая кислота), карбонат кальция для коррекции минерально-костных нарушений, а также кетостерил, элькар, витамин В6. Продолжается заместительная почечная терапия перитонеальным диализом. Планируется трансплантация почки.
Обсуждение. Настоящее наблюдение, с нашей точки зрения, представляет собой первое не только в отечественной, но и в зарубежной нефрологической литературе описание «антительного» атипичного ГУС у ребенка – носителя множественных полиморфизмов генов свертывающей системы крови. Антитела к фактору H (анти-CFH-АТ) выявляются приблизительно у 10–15% пациентов с аГУС, преимущественно у детей в возрасте 4–17 лет. Их действие направлено против С-терминального региона (экзоны 19–20) молекулы CFH, обусловливая те же последствия, что и мутации фактора Н. Наличие этих антител связано с дефицитом фактор Н-связанных белков 1 и 3 (CFHR1 и CFHR3) вследствие мутаций соответствующих генов. Установлено, что у 90% больных анти-CFH-АТ полностью отсутствуют CFHR1 и CFHR3, что обусловлено гомозиготной делецией в генах этих протеинов [13, 14].
Почти у половины пациентов с «антительным» аГУС болезнь дебютирует диарейной продромой, чем напоминает STEC-ГУС. Именно такое начало отмечено и у нашей пациентки. Особенностью аГУС, обусловленного анти-CFH-АТ, служит частое рецидивирование заболевания (58%) вследствие накопления аутоантител, причем каждый рецидив повышает риск развития хронической болезни почек (ХБП) или терминальной почечной недостаточности (ТПН). Около 40% больных таким вариантом аГУС демонстрируют развитие ХБП, у 30% пациентов развивается ТПН, 10% умирают. В диализе нуждаются от 17 до 74% пациентов, при этом АГ служит наиболее частым симптомом «антительного» аГУС [15, 16].
Поскольку «антительный» аГУС представляет собой заболевание аутоиммунное, очевидно, что лечения СЗП, с которого начинается терапия любого аГУС, недостаточно для достижения ремиссии. В связи с этим плазмотерапию следует сочетать с иммуносупрессантами для подавления выработки анти-CFH-АТ. В литературе имеются сведения об эффективном использовании ГКС и любых цитостатических препаратов – азатиоприна, микофенолата мофетила, циклофосфамида, ритуксимаба [15, 17]. Однако в последнее время при лечении «антительного» аГУС, особенно в отношении детей, все чаще стали применять экулизума [9, 18, 19]. Препарат в качестве стартовой терапии при этой форме аГУС рекомендуют назначать пациентам с тяжелым поражением головного мозга и сердца, при противопоказаниях к иммуносупрессивной терапии и отсутствии эффекта от нее, при невозможности проведения и неэффективности плазмотерапии, а также при сочетании антительного аГУС с мутациями белков – регуляторов комплемента [20]. При этом следует помнить, что экулизумаб не ингибирует продукцию анти-CFH-антител и спонтанное их снижение происходит редко [9, 15, 19].
Тяжелое состояние нашей пациентки, наличие у нее признаков поражения ЦНС (эпизоды клонико-тонических судорог), желудочно-кишечного тракта, печени, крайне высокий уровень анти-CFH-АТ, рецидивирующее течение заболевания, несмотря на лечение СЗП и преднизолоном, обосновывали назначение экулизумаба. Мы надеялись, что своевременное начало терапии и, следовательно, быстрая блокада активации комплемента позволят прекратить генерализованное микроциркуляторное тромбообразование, достичь гематологической ремиссии, восстановить функцию почек и купировать экстраренальные проявления болезни. В течение 6 месяцев лечения все цели, за исключением нормализации или хотя бы улучшения почечной функции, были достигнуты. Отсутствие положительной динамики со стороны функции почек при раннем начале терапии экулизумабом, приведшей к купированию микроангиопатической гемолитической анемии и тромбоцитопении, до настоящего времени не описано в литературе. Это привело нас к заключению о возможном наличии у ребенка генетической тромбофилии, которая могла бы через активацию локальной внутриклубочковой гиперкоагуляции усугубить ишемическое повреждение почек. Ранее нами доказано влияние носительства множественных полиморфизмов генов гемостаза на выраженность клинических проявлений (в т.ч. тяжесть ОПП) типичного ГУС у детей [21]. Аналогичную возможность мы предположили и для аГУС. Исследование полиморфизма генов свертывающей системы крови выявило у нашей пациентки сочетание 3 гомозиготных и 3 гетерозиготных мутаций практически во всех звеньях гемостаза – плазменном (фибриноген), фибринолизе (ПАИ1) и сосудисто-тромбоцитарном (фолатный цикл и тромбоцитарные рецепторы), результатом чего, с нашей точки зрения, стал дисбаланс между анти- и прокоагулянтными механизмами с преобладанием последних во внутрипочечной циркуляции. Таким образом, избыточное свертывание крови, обусловленное тромбофилией, у ребенка с аГУС могло усилить процессы тромбообразования в микроциркуляторном русле почек, вызванного неконтролируемой активацией комплемента, т.е. имел место неадекватный гемостатический ответ на болезнь, как и в случае сочетания тромбофилии с гломерулярной патологией [22]. По-видимому, комплемент-опосредованное повреждение эндотелия клубочков при аГУС у пациентов, являющихся носителями множественных полиморфизмов генов гемостаза, вызывает в микроциркуляторном русле почек локальную избыточную, «неадекватную запросу», активацию внутрисосудистого свертывания крови, подавить которую не всегда возможно даже после блокады комплемента экулизумабом (т.е. после прекращения воздействия патологического фактора), о чем свидетельствует пример нашей пациентки. Косвенным аргументом в пользу этого предположения служит быстрое нарастание диуреза с практически двукратным увеличением его объема, отмеченное у девочки через несколько дней после назначения НМГ. К сожалению, позднее начало терапии антикоагулянтами уже не смогло существенно улучшить функцию почек и ребенок остался диализзависимым.
Очевидно, что носительство множественных полиморфизмов генов гемостаза служит фактором, предрасполагающим не только к венозным и артериальным тромбозам, но и к тромбозам микроциркуляторным и в ряде случаев, вероятно, может играть роль локального триггерного механизма при любой ТМА, в т.ч. и при аГУС. Подобное предположение основано на тесной связи систем комплемента и гемостаза, которая стала изучаться в последнее десятилетие. Установлено, что компоненты каскада коагуляции оказывают модулирующее влияние на активацию комплемента. Оказалось, что тромбин обладает активностью С5-конвертазы, вызывая образование анафилотоксина С5а в отсутствие С3, что позволяет предполагать существование тромбин-зависимого пути активации комплемента. Помимо тромбина активировать систему комплемента могут некоторые факторы плазменного каскада коагуляции – IХа, Xа, XIа, а также плазмин [8, 10–12]. Кроме того, в недавнем исследовании у небольшого числа пациентов с аГУС были идентифицированы мутации гена плазминогена [23]. Эти данные позволяют объяснить тяжесть почечного повреждения у нашей пациентки и неполную эффективность экулизумаба, несмотря на своевременно установленный диагноз аГУС и раннее начало лечения. Мы полагаем, что избыточная активация комплемента у ребенка – носителя множественных полиморфизмов генов гемостаза могла сопровождаться чрезмерной гиперкоагуляцией во внутрипочечном сосудистом русле. При этом активированные факторы каскада коагуляции и в особенности тромбин, который в избытке образуется при активации внутрисосудистого свертывания крови, возможно, в свою очередь способствовали дополнительной активации системы комплемента и поддерживали ее, что привело к формированию порочного круга непрерывной и взаимоподдерживающей активации систем гемокоагуляции и комплемента.
Таким образом, сочетание у нашей пациентки неконтролируемой активации альтернативного пути комплемента, обусловленной присутствием анти-CFH-АТ в высоком титре, с избыточной внутрисосудистой активацией свертывания крови вследствие носительства множественных полиморфизмов генов гемостаза, по-видимому, привело к развитию тяжелой почечной ТМА, которую не удалось купировать экулизумабом, несмотря на то что гематологические и экстраренальные симптомы аГУС в ходе терапии регрессировали.
Заключение. Особенности течения «аутоиммунного» аГУС в представленном наблюдении, в т.ч. и ответ на терапию ингибитором комплемента экулизумабом с учетом ее своевременного начала, еще раз иллюстрируют справедливость гипотезы «двойного удара» (трансформировавшейся сегодня в гипотезу «множественных воздействий»), объясняющей развитие любого тромбоза, включая микроциркуляторного, сочетанием нескольких факторов, один из которых служит предрасполагающим, а другой (или другие) рассматривается как локальные триггеры и дополнительные механизмы, поддерживающие и усиливающие тромбообразование. Представляется обоснованным применение пациентами с аГУС и генетической тромбофилией без выраженной тромбоцитопении гепаринов (как низкомолекулярных, так и, особенно в момент острого эпизода, нефракционированного) в сочетании не только с СЗП, но и с экулизумабом.


