
Введение
IgA-нефропатия (ИГАН) – иммунокомплексный гломерулонефрит, характеризующийся преимущественным отложением в мезангии иммуноглобулина А (IgA) и мезангиальной пролиферацией [1]. ИГАН представляет собой самый распространенный вариант первичного гломерулонефрита в мире: заболеваемость им составляет 2,5 случая на 100 тыс. взрослых в год [2]. В своем классическом варианте ИГАН рассматривается как доброкачественное заболевание, редко приводящее к нарушению функции почек: с момента верификации морфологического диагноза 10- и 20-летняя почечная выживаемость составляет 95,8 и 86,1% соответственно [3]. Однако в клинической практике все чаще встречается прогрессирующее течение ИГАН, проявляющееся быстрым развитием тяжелой артериальной гипертензии и ранним нарушением функции почек; причины точно не установлены. При морфологическом исследовании почек в подобных случаях чаще всего наблюдается сочетание ИГАН и вторичного фокальносегментарного гломерулосклероза (ФСГС), а также выявляются выраженные сосудистые изменения вплоть до признаков классической тромботической микроангиопатии (ТМА).
Демонстрируем собственное клиническое наблюдение сочетания ИГАН с ТМА.
Пациент Т. 33 лет.
Анамнез жизни: курильщик, стаж курения – 20 лет, индекс курения – 19 пачка/лет. В возрасте 6 месяцев перенес флегмону правой голени. В 13 лет оперирован по поводу остеомиелита малоберцовой кости. Наследственный анамнез по сердечно-сосудистым катастрофам не отягощен.
 Анамнез morbi: с 30 лет периодически беспокоили головные боли, тянущие боли в поясничной области, по поводу чего не обследовался, артериальное давление не измерял. Однако в связи с длительным существованием жалоб в начале 2006 г. (31 год) обратился к врачу. При обследовании выявлены изменения в анализе мочи (ПУ – 1,7 г/л, ЭУ – 20–25 в п/зр) и повышение уровня сывороточного креатинина (Скр – 1,5 мг/дл). Тем не менее и в тот момент АД измерено не было. В специализированном нефрологическом отделении не наблюдался, за лечением обратился к гомеопату.
Анамнез morbi: с 30 лет периодически беспокоили головные боли, тянущие боли в поясничной области, по поводу чего не обследовался, артериальное давление не измерял. Однако в связи с длительным существованием жалоб в начале 2006 г. (31 год) обратился к врачу. При обследовании выявлены изменения в анализе мочи (ПУ – 1,7 г/л, ЭУ – 20–25 в п/зр) и повышение уровня сывороточного креатинина (Скр – 1,5 мг/дл). Тем не менее и в тот момент АД измерено не было. В специализированном нефрологическом отделении не наблюдался, за лечением обратился к гомеопату.
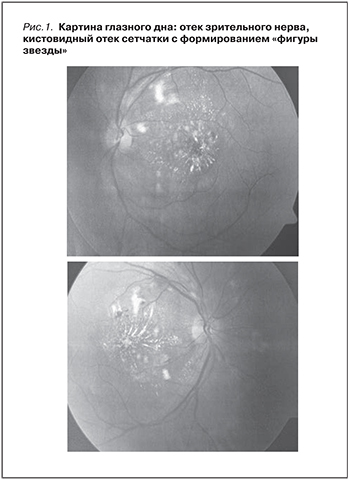
В декабре 2007 г. при усилении головных болей отметил внезапное ухудшение зрения (помутнение, диплопия, фотопсии). Обратился в клинику глазных болезней, где при первом измерении артериального давления зафиксированы цифры 240/180 мм рт.ст. с характерной картиной глазного дна (отек диска зрительного нерва, кистовидный отек сетчатки с формированием «фигуры звезды») (рис. 1). Диагностирована злокачественная артериальная гипертензия (ЗАГ), при обследовании по поводу которой выявлены анемия (Hb – 117 г/л) с нормальным числом тромбоцитов, увеличение СОЭ (50 мм/ч), выраженный мочевой синдром (ПУ 2,3 г/л, ЭУ 4–5 в п/зр) и повышение сывороточного креатинина до 4,0 мг/дл с быстрым нарастанием до 5,2 мг/дл. По данным проведенного на тот момент УЗИ и МСКТ, размеры почек были в пределах нормы, толщина паренхимы – 20–17 мм. В результате проводимой антигипертензивной (атенолол, нифедипин), диуретической терапии (фуросемид) удалось стабилизировать АД на уровне 140/80 мм рт.ст., что в сочетании с местной терапией дексаметазоном привело к восстановлению зрения и улучшению состояния глазного дна.
Однако причина развития у молодого пациента столь тяжелой АГ с поражением почек оставалась неясной. Для уточнения диагноза и определения дальнейшей тактики лечения был госпитализирован в клинику им. Е.М. Тареева в апреле 2008 г.
При поступлении: состояние средней тяжести, жалобы на тошноту, кожный зуд, сонливость, общую слабость, снижение аппетита. Рост – 180 см, вес – 85 кг, при аускультации легких дыхание везикулярное, хрипов нет, ЧД – 16 уд./мин, Тоны сердца ритмичные, пульс 72 – уд/мин, АД – 150/100 мм рт.ст. Живот мягкий безболезненный. Периферических отеков нет. Печень и почки не пальпируются. Симптом поколачивания отрицательный с обеих сторон.
При обследовании сохранялись выраженный мочевой синдром (СПУ – 1,7 г/сут), гематурия (ЭУ – густо в п/зр), стабильно сниженная функция почек (Скр – 5,4 мг/дл, по данным пробы Реберга, СКФ – 23 мл/мин), анемия с продолжающимся снижением уровня гемоглобина до 112 г/л при нормальном числе тромбоцитов. При обследовании маркеры вирусных гепатитов В и С, ВИЧ и сифилиса отрицательные. Данные за наличие системных заболеваний отсутствовали (АНФ – отр., АТ к ДНК – отр., РФ – отр., комплемент в пределах нормальных значений, СРБ – отр., криоглобулины – отр.). Были также исключены амилоидоз, миеломная болезнь и парапротеинемии. Впервые обнаруженное повышение уровня IgA сыворотки до 350 мг/дл (норма – 50–300 мг/дл) в сочетании с выраженной микрогематурией давало основание обсуждать диагноз ИГАН. Быстрое прогрессирование заболевания, ЗАГ, причины которой оставались неясными, а также признаки гиперкоагуляции с повышением уровня Д-димеров (Д-димеры более 3 мкг/мл при норме до 05 мкг/мл) требовали исключения тромбофилии. При обследовании на антифосфолипидный синдром (АФС) обнаружен циркулирующий волчаночный антикоагулянт (ВА). Генетическая тромбофилия была подтверждена обнаружением полиморфизмов генов PAI-1 (генотип 4G/4G), β-цепи фибриногена (генотип А/А) и MTHFR677 (генотип С/Т).
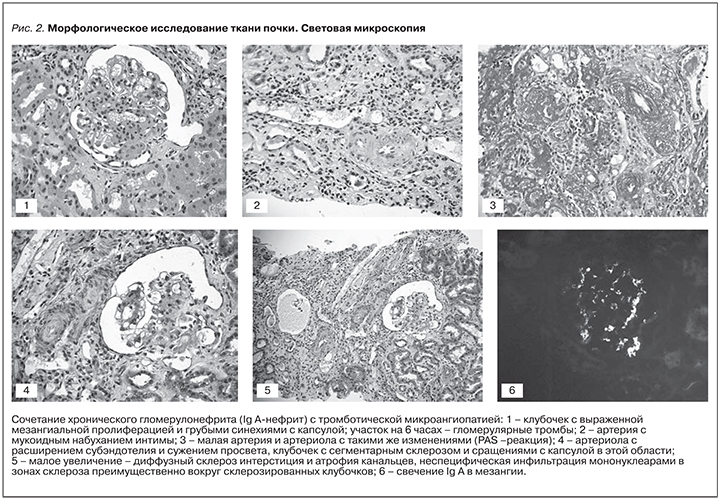
Для уточнения характера поражения почек выполнена нефробиопсия (проведены окраски: Г-Э, ШИК-реакция, трихром по Массону): в препарате 6 клубочков, 1 из них полностью склерозирован; в оставшихся клубочках отмечается выраженное расширение мезангия и пролиферация мезангиоцитов; в 2 клубочках имеются участки сегментарного склероза и сращения с капсулой Боумена в этих областях; диффузный склероз интерстиция и атрофия канальцев; неспецифическая инфильтрация мононуклеарами в зонах склероза, преимущественно вокруг склерозированных клубочков; артерии – явления миоинтимальной пролиферации в артериях малого калибра; артериолы – утолщение стенок артериол за счет мукоидного набухания. Иммунофлюоресценция: IgG – отрицательно, IgA – мезангий +++, IgM – отрицательно, С3 – то же, что IgA, фибрин – эндотелий сосудов +. Заключение: IgA-нефропатия (II класс по классификации M. Haas). Тромботическая микроангиопатия (рис. 2).
Таким образом, гистологическая картина в сочетании с данными клинико-лабораторного обследования позволила диагностировать ИГАН в сочетании с ТМА, которая, по-видимому, определяла быстрое прогрессирование заболевания.
С учетом признаков активности гломерулонефрита была начата активная иммуносупрессивная терапия: метипред 20 мг/сут per os, сочетанные «пульсы» ПЗ и ЦФА (суммарная доза – 3000 и 1200 мг соответственно). С целью коррекции анемии назначены препараты эритропоэтина и железа, для коррекции АД – атенолол 50 мг/сут, фелодипин 10 мг/сут, диуретическая терапия – фуросемид 40 мг через день. Несмотря на наличие гиперкоагуляции, назначение НМГ ограничивали низкие показатели СКФ. Для профилактики дальнейшего микротромбообразования к терапии добавлен тромбо-асс 50 мг/сут. В результате проведенной терапии отмечено снижение уровня сывороточного креатинина с 5,4 до 4,4 мг/дл, уменьшилась выраженность мочевого синдрома (СПУ – 0,96 г/ сут, ЭУ – 2–3 в п/зр), нормализовался уровень гемоглобина (Hb – 126 г/л), АД стабилизировано на уровне 120–130/85–90 мм рт.ст. Начато постепенное снижение пероральной дозы метипреда до 12 мг/сут, проведен еще один сочетанный «пульс» ПЗ и ЦФА (1500 и 600 мг соответственно). Самочувствие больного значительно улучшилось, уменьшилась слабость, исчезла тошнота.
В течение одного месяца, до начала июня 2008 г., АД сохранялось на достигнутом уровне, азотемия не нарастала (Скр – 4,2 мг/дл, СКФ – 20 мл/мин), выраженность мочевого синдрома была небольшой (СПУ – 0,52 г/сут, ЭУ – 10–12 в п/зр).
Однако у пациента появились эпизоды нарушения ритма сердца (ощущение сильного сердцебиения, перебоев в работе сердца), частота и продолжительность которых нарастали, что потребовало повторной госпитализации в клинику им. Е.М. Тареева. По данным коагулограммы, вновь выявлены признаки гиперкоагуляции (фибриноген – 4,3 г/л, протромбиновый индекс – 117%, Д-димеры – 0,5–3,0 мкг/мл [N<0,5]); волчаночный антикоагулянт не обнаружен. По данным УЗИ отмечено уменьшение размеров почек (ЛП – 87×42 мм, ПП – 87×47 мм, толщина паренхимы – 10–11 мм).
При проведении холтеровского мониторирования ЭКГ были выявлены эпизоды предсердной тахикардии, в т.ч. мерцания – трепетания предсердий (9 продолжительностью до 22 с, ЧСС – 178/мин, R-R – 350 мс, SТ-Т без диагностически значимой динамики).
 Для уточнения характера кардиальной патологии выполнено ЭхоКГ: толщина стенок ЛЖ: МЖП – 0,9 см, ЗС – 1,0 см; глобальная сократительная функция ЛЖ не нарушена, нарушений локальной сократимости нет. Диастолическая функция не нарушена. ЛП – 40 мл, ПП – 34 мл, межжелудочковая перегородка без особенностей. Клапанный аппарат не изменен. Диаметр корня аорты – 3,3 см. В связи с отсутствием сердечной недостаточности, по данным ЭхоКГ, для исключения возможного поражения интрамиокардиальных сосудов в рамках ТМА как возможной причины развития аритмии пациенту была выполнена перфузионная сцинтиграфия миокарда. При проведении исследования отчетливо визуализировалось включение индикатора в миокард ЛЖ, полость желудочка незначительно расширена, визуальные признаки гипертрофии миокарда не выявлены; распределение радиофармпрепарата в стенке желудочка диффузное, неравномерное, с множественными мелкими полиморфными дефектами аккумуляции практически во всех отделах миокарда (передняя, нижняя, боковая стенки, верхушка) без четкой преимущественной локализации; включение индикатора в миокард правого желудочка незначительное (пренебрежительно мало). Заключение: признаки диссеминированного некоронарного поражения миокарда.
Для уточнения характера кардиальной патологии выполнено ЭхоКГ: толщина стенок ЛЖ: МЖП – 0,9 см, ЗС – 1,0 см; глобальная сократительная функция ЛЖ не нарушена, нарушений локальной сократимости нет. Диастолическая функция не нарушена. ЛП – 40 мл, ПП – 34 мл, межжелудочковая перегородка без особенностей. Клапанный аппарат не изменен. Диаметр корня аорты – 3,3 см. В связи с отсутствием сердечной недостаточности, по данным ЭхоКГ, для исключения возможного поражения интрамиокардиальных сосудов в рамках ТМА как возможной причины развития аритмии пациенту была выполнена перфузионная сцинтиграфия миокарда. При проведении исследования отчетливо визуализировалось включение индикатора в миокард ЛЖ, полость желудочка незначительно расширена, визуальные признаки гипертрофии миокарда не выявлены; распределение радиофармпрепарата в стенке желудочка диффузное, неравномерное, с множественными мелкими полиморфными дефектами аккумуляции практически во всех отделах миокарда (передняя, нижняя, боковая стенки, верхушка) без четкой преимущественной локализации; включение индикатора в миокард правого желудочка незначительное (пренебрежительно мало). Заключение: признаки диссеминированного некоронарного поражения миокарда.
Поражение сердца, расцененное как экстраренальная ТМА, стало показанием к назначению низкомолекулярных гепаринов, несмотря на низкие показатели СКФ. Терапия эноксапарином в дозе 40 мг/сут в течение 2 недель привела к прекращению приступов тахиаритмии.
В дальнейшем, несмотря на проведенную имммуносупрессивную, антикоагулянтную и нефропротективную терапию, пациент достиг 5-й стадии ХБП и после формирования артериовенозной фистулы в плановом порядке была начата заместительная почечная терапия (ЗПТ) программным гемодиализом, в связи с чем иммуносупрессивная терапия прекращена. После нескольких лет заместительной почечной терапии пациенту была выполнена аллотрансплантация почки. Отмечена отсроченная функция трансплантата, однако при добавлении к терапии гепарина функция органа нормализовалась.
Обсуждение
Особенности представленного наблюдения: сочетание гистологической картины ИГАН с признаками ТМА, злокачественная АГ и темп прогрессирования почечной недостаточности (от момента дебюта заболевания до начала ЗПТ – 3 года), не характерные для данного морфологического варианта гломерулонефрита.
Можно предположить, что именно присоединение ТМА к имеющемуся иммуновоспалительному процессу в клубочках почки послужило причиной бурной манифестации болезни с быстрым достижением терминальной почечной недостаточности. Необходимо отметить особенности ТМА у нашего пациента, как морфологические, так и клинические. Первые заключались в преимущественно артериолярной локализации повреждения и отсутствии тромбозов при светооптической микроскопии. Однако выявление депозитов фибрина в эндотелии сосудов при ИФМ свидетельствует в пользу наличия фибриновых тромбов в микроциркуляторном сосудистом русле. К клиническим особенностям ТМА в данном случае можно отнести ее генерализованный характер в отсутствие гематологических проявлений, поскольку анемию, имевшуюся у пациента, при наличии стойко нормального числа тромбоцитов и быстрого положительного ответа на терапию эритропоэтинами скорее всего следует рассматривать как нефрогенную, а не как микроангиопатическую, хотя маркеры микроангиопатического гемолиза тогда не исследовались.
Можно предполагать, что у нашего пациента ЗАГ была не причиной ТМА, а ее следствием в результате ишемического повреждения почек, развившегося в результате микротромбообразования. Дополнительным аргументом в пользу этой точки зрения служит развитие экстраренальной ТМА на фоне уже стабилизированного АД и в отсутствие прогрессирования заболевания почек.
До недавнего времени считалось, что ТМА при ИГАН встречается нечасто, однако эти сведения противоречивы. По данным Nasri Hamid, гистологические признаки ТМА были выявлены лишь в 2% биоптатов больных ИГАН, причем развитие ТМА связывали с сопутствующей злокачественной артериальной гипертензией [4]. С другой стороны, El Karoui и соавт. обнаружили признаки ТМА у 53% больных ИГАН, что авторы также объясняют отчасти тяжелой артериальной гипертензией и продвинутой стадией заболевания с нарушением функции почек [5].
Причина развития ТМА при ИГАН остается не вполне ясной. В исследовании El Karoui и соавт. показано, что частота ТМА, ассоциированной с ИГАН, заметно возрастает с увеличением выраженности артериальной гипертензии. Это дает основание полагать, что повышение артериального давления служит фактором риска ТМА. Однако кажется маловероятным, что тяжелая АГ может быть единственной причиной ТМА, ассоциированной с ИГАН, т.к. в 29% случаев признаки ТМА были выявлены у пациентов с систолическим давлением <140 мм рт.ст. на время проведения биопсии. Также не было обнаружено связи ТМА с выраженностью интерстициального фиброза и повреждения канальцев. Из этого следует, что ТМА скорее предшествует развитию гломерулосклероза и интерстициального фиброза, а не является их следствием [5]. Вопрос, что первично – ЗАГ или ТМА, до настоящего времени дискутируется в литературе. Считается, что ТМА развивается у трети больных ЗАГ, проявляясь классическими клиническими симптомами – микроангиопатическим гемолизом, тромбоцитопенией и нарушением функции почек. Однако в последнее время все чаще наблюдается отсутствие гематологической составляющей синдрома ТМА [6] при его несомненных гистологических признаках, как это имело место у нашего пациента.
Таким образом, в данном случае более вероятной генетической причиной представляется наличие ТМА, чем ЗАГ. Выявленные у больного полиморфизмы генов системы гемостаза дают основание обсуждать связь ТМА с наследственной тромбофилией.
В последнее время в патогенезе ТМА все больше уделяется внимание гиперкоагуляционным нарушениям. Однако чаще развиваются тромбозы сосудов микроциркуляторного русла, обусловленные генетическим дефектом в системе комплемента (аГУС), приводящие к повреждению эндотелиальных клеток и нарушению сосудисто-тромбоцитарного гемостаза.
У пациентов с ТМА при ИГАН не было выявлено мутаций генов регуляторных факторов комплемента CFH, CFI и MCP, ассоциированных с аГУС [5]. В последнее время были получены данные о возможной взаимосвязи некоторых полиморфизмов генов плазменного звена коагуляции с риском развития ТМА [8–11], не исследованных при ИГАН с ТМА. Была выявлена статистически значимая ассоциация риска развития ТМА и носительства ТТ генотипа фактора XIII, приводящая к снижению фибринолитической активности [10]. В исследовании Fengxiao Bu и соавт. наибольшую значимость ассоциации с аГУС показал полиморфизм гена плазминогена (PLG) [11].
В то же время другим значимым генетическим полиморфизмом, ассоциированным с ТМА, стал ТТ генотип С677Т метилентетрогидрофолатредуктазы, что, возможно, объясняется активацией эндотелия и повышенным оксидативным стрессом на фоне гипергомоцистеинемии [12]. Наиболее часто тестируются мутации фактора V. Лейдена (FVL) и протромбина G20210A, при которых не выявлено ассоциации с развитием ТМА [8], однако при этом не исследуются другие полиморфизмы генов гемостаза, а тем более возможное сочетанное действие нескольких полиморфизмов.
Уже длительное время существует концепция полигенного характера наследственной тромбофилии, согласно которой генетическая предрасположенность к тромбозу обусловлена не повреждением отдельного гена, а комбинацией нескольких генетических дефектов. Таким образом, особое значение в развитии ТМА может иметь сочетание нескольких полиморфизмов генов коагуляции, что доказали в своем исследовании Л.А. Боброва и соавт., по результатам которого у больных хроническим гломерулонефритом (ХГН) независимо от его морфологического варианта при наличии мультигенной формы тромбофилии в 30% случаев отмечается развитие острой ТМА [13].
Таким образом, полагаем, что мультигенная форма тромбофилии может играть самостоятельную роль в развитии ТМА у больных ИГАН, причем не только почечной, но и экстраренальной.
В начале наблюдения за нашим больным тромбофилия рассматривалась как комбинированная в связи с выявлением не только генетических маркеров, но и циркулирующего волчаночного антикоагулянта. Однако отсутствие у пациента тромбозов крупных сосудов и лишь однократное обнаружение антифосфолипидных антител позволили исключить АФС как дополнительную причину тромбофилии.
Можно возразить, что у данного пациента не было выявлено наиболее «протромбогенных» полиморморфизмов генов системы гемостаза, а общее количество полиморфизмов не так велико. Однако стоит отметить, что ТМА, в итоге приобретшая системный характер, развилась у больного с выраженной активностью нефрита, не только клинической, но и морфологической. Связи системы воспаления и коагуляции прочны и многоуровневы. Активация одной системы незамедлительно приводит к активации второй. Применительно к хроническому гломерулонефриту это означает активацию внутрисосудистого свертывания крови в гломерулярных и мелких экстрагломерулярных сосудах.
В пользу гиперкоагуляции в патогенезе ТМА у данного пациента свидетельствует постоянная активация внутрисосудистого свертывания по данным лабораторным сведений и быстрое купирование клинических проявлений ТМА миокарда и восстановление функции трансплантата при добавлении к терапии НМГ.
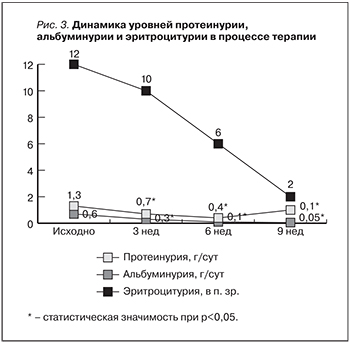
Таким образом, полагаем следующий ход событий: активация иммунновоспалительного процесса в рамках ИГАН влечет за собой избыточную активацию коагуляции у пациента с тромбофилией, что в свою очередь индуцирует микротромбообразование в микроциркуляторном русле почек с развитием в дальнейшем ишемического поражения. ЗАГ в данной ситуации можно рассматривать как следствие ишемии. Причем каждая из упомянутых видов патологии будет способствовать прогрессированию почечной недостаточности (рис. 3).
Наше предположение оправданно также тем, что мультигенная тромбофилия сама по себе способствует развитию нефросклероза [14], вероятно, через развитие ишемии почечной ткани, вызванной тромбообразованием в сосудистом русле почек, в первую очередь в мелких сосудах, обеспечивающих перфузию интерстиция, что также могло способствовать быстрому прогрессированию почечной недостаточности.
Неэффективность лечения данного пациента стала следствием поздней диагностики заболевания, обращением за медицинской помощью на уже продвинутой стадии почечной недостаточности. Можно предположить, что при условии более ранней диагностики полноценная иммуносупрессивная терапия в сочетании с НМГ могла бы затормозить темп прогрессирования заболевания, тем более что назначение НМГ в какой-то степени позволило отсрочить наступление терминальной стадии почечной недостаточности.
Заключение
Данное клиническое наблюдение подтверждает возможность тяжелого течения и быстрого прогрессирования ИГАН – как правило, доброкачественного заболевания при сочетании его с ТМА. Пример нашего больного в сочетании с имеющимися данными о нарастании частоты развития ТМА при ИГАН заставляют проявлять настороженность при нетипичном течении заболевания. В таких ситуациях необходимо включать в спектр лабораторной диагностики расширенную коагулограмму и при выявлении активации свертывающей системы исключать возможные причины тромбофилии, в т.ч. и генетические. В этих случаях биопсия почки особенно необходима, поскольку она позволяет объяснить те предположения, которые у нас возникают при клиническом наблюдении за больным. Положительный эффект от назначения НМГ при почечной ТМА подтверждается множеством клинических примеров, надеемся, в ближайшем будущем оно войдет в широкую практику.


