
Введение
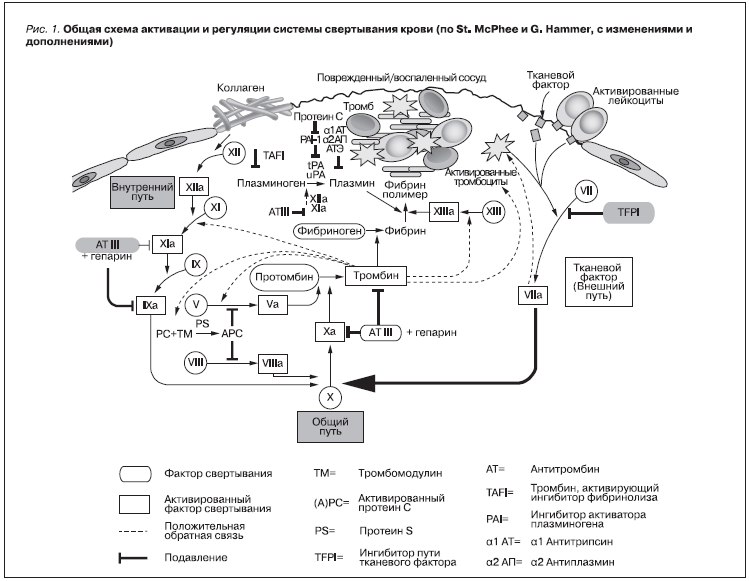
Система гемостаза – биологическая, обеспечивающая, с одной стороны, сохранение жидкого состояния циркулирующей крови, с другой – предупреждение и купирование кровотечений [1, 2]. На рис. 1 представлен комплекс про- и антикоагуляционных механизмов, приводящих к образованию гемостатической пробки и восстановлению сосудистой стенки [3]. Изменения в системе гемостаза могут приводить к патологии органов мочевой системы, в первую очередь к развитию тромбозов сосудистого русла почек, в т. ч. почечных вен, артерий и поражение микроциркуляторного русла.
Тромбоз почечных вен (ТПВ) представляет собой редкую форму тромботического поражения сосудов почек, развивающуюся при сочетании приобретенных и наследственных протромботических факторов риска [4, 5]. У новорожденных до 40 % всех эпизодов венозной тромбоэмболии представлено именно ТПВ [6], общая встречаемость которого у детей, находящихся в отделениях интенсивной терапии, составляет 0,5 на 1000. У большинства (75 %) новорожденных ТПВ являются односторонним процессом [5], двусторонние поражения встречаются чаще у недоношенных и маловесных детей (до 1500 г) [7]. Преимущественно страдают мелкие интраренальные вены – дуговые или междольковые, реже – более крупные междоле-
вые и нижняя полая вены. ТПВ проявляется минимальной гематурией, симптомом “пальпируемой опухоли” в животе, тромбоцитопенией, артериальной гипертензией, нарушением функции почек [8]. Диагноз подтверждается при проведении ультразвукового исследования почек (УЗИ) с допплерографией (УЗДГ) сосудов почек, выявляющих снижение амплитуды или полное отсутствие венозного сигнала, а также структурные изменения почек – сначала увеличение в размерах с потерей
кортико-медуллярной дифференцировки, а по прошествии острой фазы – постепенное их сморщивание. В ранние сроки после тромбоза при использовании высокочастотных датчиков
выявляются гиперэхогенные полоски [9–11], идущие сквозь паренхиму почки, – это мелкие тромбированные сосуды (интерлобарные и интерлобулярные вены). По данным S. Marks и соавт. (2005), в исходе ТПВ у 34 % детей (13 из 43) развилась артериальная гипертензия, у 29 % (11 из 43) – сморщивание почек и почечная недостаточность [12], что согласуется с результатами других исследователей [9].
Поражение артериального русла почек может быть представлено стенозом почечной артерии (т. н. ишемической нефропатией – медленнопрогрессирующим заболеванием, чаще связанным с атеросклеротическими изменениями или фиброзно-мышечной дисплазией и проявляющимся артери-
альной гипертензией) [13, 14] и тромбозом артерий (инфаркт почки – ИП).
Под тромбофилией понимают предрасположенность организма к развитию тромбозов вследствие нарушения регуляторных механизмов системы гемостаза или изменения свойств отдельных ее звеньев [15,16]. Термин “тромбофилия” используется для обозначения как наследственных, так и приобретенных причин повышенного тромбообразования. К наследственным причинам относят дефицит естественных антикоагулянтов (антитромбина III, протеинов С и S), мутацию G1691A в гене V фактора свертывания (Лейденская мутация), мутацию G20210A в гене протромбина, мутацию С677Т в
гене метилентетрагидрофолатредуктазы (MTHFR), а также некоторые другие полиморфизмы генов гемостаза, которых к настоящему времени уже выявлено несколько десятков [17–20]. У пациентов с наследственной тромбофилией тромбозы часто обусловлены сочетанием нескольких генных полиморфизмов системы гемостаза [21–24]. Н.А. Макацария (2010) критериями наследственной тромбофилии предлагает считать мутации фактора V и гена протромбина, дефицит естественных антикоагулянтов, носительство трех любых гомозиготных полиморфизмов, либо пяти гетерозиготных, а также умеренную и тяжелую гипергомоцистеинемию [25].
Рисунок 1. Общая схема активации и регуляции системы свертывания крови (по St. McPhee и G. Hammer
с изменениями и дополнениями).
Исследования последних лет существенно расширили наши представления о молекулярных механизмах формирования тромбофилических состояний. К настоящему времени выявлено несколько десятков генетических дефектов, имеющих прямое или опосредованное отношение к нарушениям гемостаза,
наличие которых ассоциировано с развитием протромботических сдвигов и/или риском тромбозов. Большинство из этих генов кодирует компоненты плазменного и тромбоцитарного звеньев гемостаза либо предрасполагает к развитию состояний, вовлеченных в патогенез эндотелиальной дисфункции
(гипергомоцистеинемия, гиперлипопротеинемия, артериальная гипертензия и др.). Однако, несмотря на общепризнанное мнение о протромботической роли указанных аномалий, по-прежнему существует множество вопросов относительно их участия в патогенезе развития артериальных/венозных тромбозов и целесообразности диагностики этих мутаций в практических целях [26–28]. Сложившаяся ситуация во многом объясняется многофакторной природой тромбозов в каждом случае и сложным характером взаимодействия генетических и экзогенных факторов риска, лежащих в основе или провоцирующих развитие патологических сдвигов в системе гемостаза.
С начала XXI в. некоторые ученые ставили вопрос о необходимости введения скрининга всего населения на наследственную тромбофилию, сравнимого с неонатальным скринингом на нарушение обмена веществ [29, 30]. Однако проводить такой скрининг современными исследователями не рекомендуется: частота тромбозов в детском возрасте слишком мала, терапия заболевания “тромбоз” вполне осуществима, а затраты, связанные с общим скринингом, весьма высоки [31, 32]. Некоторые
авторы предлагают проводить обследование всех женщин на наличие протромбогенных мутаций перед началом приема оральных контрацептивов [33, 34], хотя и это экономически не выгодно. J. Dalen, J. Dietrich предлагают обследовать только женщин из семей с повышенным риском тромбозов [35, 36]. Остается спорным и вопрос о необходимости проведения исследования родственников пациента с тромбозом при подтверждении у него генотипических сочетаний, ассоциированных с риском тромбозов [37]. В настоящее время не вызывает сомнений необходимость обследования родственников первой линии ребенка с симптомами заболевания (в первую очередь родителей и сибсов) [38, 39]; в случае обратной ситуации (болен взрослый) существуют разные мнения: E. Harris,
I. Martinelli полагают, что в этом необходимости нет [40, 41]. По мнению других авторов, сегодня имеются все возможности по проведению действенной профилактики тромбозов, в связи с чем дети из таких семей должны быть обследованы [42–44]. Некоторые исследователи ставят под сомнение необходимость проведения генетического обследования взрослых пациентов с первым эпизодом венозного тромбоза [45]. В отношении детского населения считается, что любой эпизод артериального/венозного тромбоза требует обследования ребенка на наличие генотипических сочетаний, ассоциированных с риском тромбозов [46]. Следовательно, необходим индивидуальный подход в каждом конкретном случае [47].
Среди опубликованных работ встречаются единичные описания ИП у детей, как правило, связанного с тромбированием места анастомоза после проведения трансплантации почки [48, 49]. У новорожденных детей тромбоз почечной артерии развивается как осложнение катетеризации пупочной артерии [50]. В исследованиях О.Л. Чугуновой и соавт. (2003) показано, что новорожденные с ИП имеют выраженные гипоксически-ишемические нарушения на фоне морфофункциональной
незрелости органов и систем, а также отечный синдром, склонность к урежению мочеиспусканий, увеличение почек в размерах и развитие неолигурической острой почечной недостаточности [51].
Случаи ранних тромботических эпизодов уже не являются казуистикой. Развитие у ребенка тромбоза любой локализации может приводить к тяжелым последствиям, инвалидизации и даже летальному исходу, что диктует необходимость своевременной диагностики данного состояния и по возможности
поиска мер профилактики. Дети с наследственной тромбофилией наиболее уязвимы в плане развития у них тромбозов сосудов различных органов, в т. ч. почек. Клинические проявления ИП зависят от его размеров. Небольшие инфаркты, затрагивающие часть коркового вещества почек, не имеют значимых симптомов. Более крупные инфаркты могут вызывать картину “острого живота” или маскироваться симптомами почечной колики [52, 53].
В последнее время появляются работы, подтверждающие генетическую основу ИП (наличие генотипических сочетаний, ассоциированных с риском тромбозов) у взрослых пациентов [54, 55, 56]. Практически не встречается исследований, посвященных прижизненной диагностике ИП у детей, не выделены группы риска, угрожаемые по его развитию. Целью данного исследования было определить критерии диагностики ИП у детей и выделить группу риска по развитию тромбозов сосудов почек у детей с наследственной тромбофилией.
Материал и методы
За 2008–2010 гг. нами обследованы 66 детей в возрасте от года до 18 лет (средний возраст (М ± SD) – 9,0 ± 5,4) с наследственной тромбофилией, разделенных на две подгруппы основной группы: 1-ю подгруппу составили дети соматически здоровые, являющиеся носителями генотипических сочетаний, ассоциированных с риском тромбозов (всего 44 ребенка), которые были обследованы в связи с отягощенным семейным анамнезом; 2-ю подгруппу – дети с наследственной тромбофилией и перенесшие острый тромбоз различной локализации (22 ребенка). Группу контроля составили 20
детей с нейрогенной дисфункцией мочевого пузыря и ночным моносимптомным энурезом, наблюдавшихся на базе ДГКБ № 13 им. Н.Ф. Филатова.
В первой подгруппе наблюдались дети от матерей, страдавших артериальной гипертензией и хроническим гломерулонефритом, либо перенесших острый тромбоз различной локализации (чаще – вен нижних конечностей), проходивших лечение в клинике нефрологии, внутренних и профессиональных болезней им. Е.М. Тареева Университетской клинической больницы № 3 Первого ГМУ им. Сеченова и ГКБ № 1 соответственно. Все женщины имели наследственную тромбофилию.
Во вторую подгруппу включены дети, наблюдавшиеся на базе гематологического консультативного отделения Измайловской детской городской клинической больницы врачом-гематологом П.В. Свириным в связи с перенесенным острым тромбозом различной локализации, в основном ишемическим инсультом
(64 % перенесли инсульт).
Группы детей были сравнимыми: средний возраст (М ± SD) в 1-й подгруппе составил 9,32 ± 5,41 года, во 2-й подгруппе – 8,45 ± 5,5 года; в группе контроля – 10,3 ± 5,0 лет. В обеих подгруппах основной группы преобладали мальчики (в 1-й подгруппе – 65,9 %, во 2-й – 63,6 %), в группе контроля отмечено равное распределение по полу.
Дети первой подгруппы прошли генетическое обследование на наличие шести генотипических сочетаний, ассоциированных с риском тромбозов, на базе лаборатории молекулярной генетики человека НИИ молекулярной медицины ГОУ ВПО Первого МГМУ им. И.М. Сеченова (зав. лаб. – проф., д.б.н. Д.В. Залетаев). Дети второй подгруппы были обследованы до включения в исследование в лаборатории ЗАО ПИННИ. Определены полиморфизмы следующих генов: протромбина (G20210A), фактора V (Лейденская мутация) (G506A), метилентетрагидрофолатредуктазы (MTHFR) (C677T), ингибитора активатора плазминогена типа 1 (PAI-I) (675: 5G del 1G), фибриногена (G455A), тромбоцитарного рецептора фибриногена (GP IIIa) (Т33ЗC).
Помимо сбора семейного анамнеза и оценки соматического статуса с антропометрией, измерением артериального давления и определением наличия мезенхимальной дисплазии всем детям осуществлено комплексное лабораторно-инструментальное обследование, включившее развернутый клинический и биохимический анализы крови и мочи, исследование активности ферментов мочи, определение микроальбуминурии полуколичественным методом с помощью тест-полосок “Микраль−Тест”; Roche. Тест считался положительным, если концентрация альбумина превышала 20 мг/л, что определялось
изменением цвета тест-полоски.
В сыворотке крови исследованы уровни гомоцистеина (иммуноферментным методом) и липопротеина (а) (методом иммунотурбидиметрии).
Для оценки скорости клубочковой фильтрации (СКФ) использовалась формула Шварца. СКФ = k × рост (см)/креатинин (мкмоль/л) × 88,4, где k для детей старше года = 0,55, для мальчиков-подростков k = 0,7; 88,4 – коэффициент перевода креатинина из мг/дл в мкмоль/л. Оценивалась коагулограмма с определением активности естественных антикоагулянтов (протеины S и C, антитромбин III). Для
исключения антифосфолипидного синдрома как приобретенного тромбофилического состояния исследовался уровень волчаночного антикоагулянта и антикардиолипиновых антител.
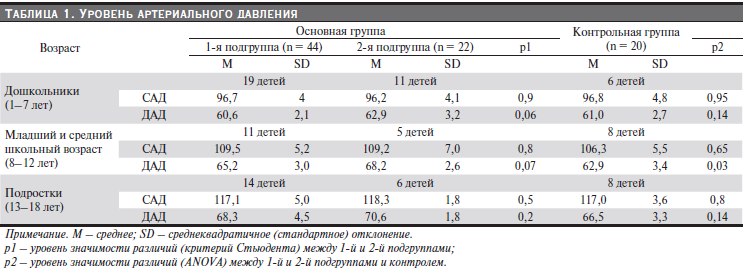
Таблица 1. Уровень артериального давления.
Ультразвуковое исследование с допплерографией сосудов почек осуществляла зав. отделением ультразвуковой диагностики КДЦ ДГКБ № 13 им. Н.Ф. Филатова д.м.н. А.И. Гуревич на аппарате Logiq-3 Expert, США; с использованием конвексного датчика 5,0 МГц и линейного датчика 8,6–11,0
МГц. Проведен осмотр почек в В-режиме, исследован кровоток при цветном допплеровском картировании (ЦДК) и дуплексном допплеровском сканировании с оценкой индекса резистентности.
Статистическая обработка результатов
Полученные данные подвергались статистической обработке при помощи программы STATISTICA 6.0. for Windows корпорации StatSoft; Russia. Анализ соответствия вида распределения признака закону нормального распределения проведен с помощью критерия Шапиро–Уилка. В зависимости от соответствия данных нормальному распределению при описании количественных данных использованы следующие расчетные показатели: Ме (IQR), где Me – медиана, IQR (interquartile range) – интерквартильный размах между значениями 25–75 процентилей (при распределении данных, отличающихся от нормального), либо М (SD), где М – среднее арифметическое, SD (Standard deviation) – стандартное отклонение (при нормальном распределении данных). Для показателей, характеризующих качественные признаки, указывались абсолютное число и относительная величина в процентах (%), а для их сравнения применяли точный критерий Фишера (двусторонний вариант). Для проверки согласия наблюдаемых и ожидаемых частот вычислен критерий хи-квадрат (χ2). Проведен расчет рисков с определением соотношения шансов (OR, Odds Ratio) и относительного риска (RR, Risk Ratio). Значение этих показателей считалось значимым, если их 95 % доверительный интервал (confidence interval (CI) не включал единицу. В целях выявления характера взаимодействия между
показателями, характеризующими изменение в организме ребенка, анализировалась ранговая корреляция Спирмена (Rs). При проведении сравнения сформированных групп пациентов для признаков, имеющих нормальное распределение, использован критерий Стьюдента (при выполнении
второго условия – равенства дисперсий, проверенного по критерию Левена), в случае множественных сравнений – параметрический однофакторный анализ вариаций (ANOVA). Для признаков, имеющих распределение, отличное от нормального, использовался критерий Манна–Уитни, в случае множественных сравнений – критерий Краскела–Уоллиса. С целью преодоления проблемы множественных сравнений применялась поправка Бонферрони. Критическое значение уровня статистической значимости при проверке нулевых гипотез принималось равным 5 % (p = 0,05).
Результаты и обсуждение
Оценку семейного анамнеза родственников осуществляли с целью выявления заболеваний, ассоциированных с артериальными/венозными тромбозами, патологией беременности у женщин, наличием заболеваний почек в семье, в частности хронического гломерулонефрита и артериальной гипертензии. Установлено, что отягощенный акушерский анамнез (случаи замершей беременности на ранних сроках, самопроизвольных выкидышей и преждевременных родов), а также тромботические эпизоды встречались одинаково часто в обеих подгруппах (в 1-й – в 82 % (31/38), во 2-й – в 77 % (17/22), двусторонний точный критерий Фишера: p = 0,74). При сравнении семейного анамнеза детей основной и контрольной групп выявлено статистически значимое различие в частоте встречаемости этих признаков: в основной группе указание на отягощенность семейного анамнеза составляло 72,7 (48/66), в контрольной группе – 20 % (4/20) (двусторонний точный критерий Фишера: p = 0,02). Наличие у матери патологии беременности (особенно повторные случаи замершей беременности на ранних сроках, самопроизвольных выкидышей), тромбозов различных локализаций, в первую очередь вен нижних конечностей и тромбоэмболии легочной артерии, следует рассматривать как возможный маркер тромбофилии у ребенка.
Оценка физического развития по центильным таблицам (А.В. Мазурин, И.М. Воронцов, 2000) показала, что 33 (75 %) ребенка 1-й подгруппы и 11 (50 %) детей 2-й подгруппы имели среднее физическое развитие. Оценка гармоничности физического развития выявила преобладание детей с гармоничным
развитием во второй группе. В первой подгруппе по сравнению со второй наблюдались дети, имеющие избыток массы тела. Статистически значимых различий по физическому развитию между подгруппами детей с наследственной тромбофилией соматически здоровых и перенесших тромбоз различной
локализации не выявлено (р = 0,5 для роста, р = 0,7 для массы тела по критерию Стьюдента).
Сопутствующая патология встречается у 71 % (47/66) детей с наследственной тромбофилией. Число патологически измененных систем и нозологических форм в пересчете на одного больного выше у детей, перенесших тромбоз: 0,8 – в первой подгруппе к 1,4 – во второй.
Уровень артериального давления, измеренный трехкратно с определением среднего АД за три измерения, не выявил артериальной гипертензии ни у одного ребенка основных групп и группы контроля (табл. 1).
Показатели АД у пациентов 1-й и 2-й подгрупп не различались между собой (p > 0,05, критерий Стьюдента).
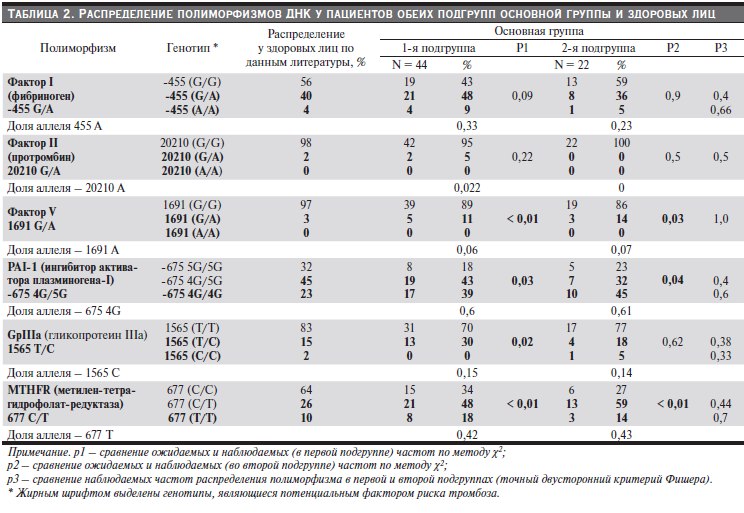
Генетические исследования проведены всем детям основной группы. В табл. 2 представлены результаты анализа распределения генотипов изученных генов у детей соматически здоровых и перенесших тромбозы различных локализаций. Статистически значимые различия между здоровыми лицами (по данным литературы) и детьми основной группы выявлены при анализе полиморфизмов в генах фактора V, MTHFR и PAI-1. Носителями варианта “1691А” гена фактора V (мутации FV Leiden) в гетерозиготном состоянии являлись 5/44 (11 %) ребенка первой подгруппы и 3/19 (14 %) детей, перенесших тромбоз, что при сравнении со здоровыми оказалось выше в 3,5–4,0 раза. Частота гомозиготного носительства полиморфизмов генов MTHFR и PAI-1 оказалась выше в 1,5 и 2,0 раза
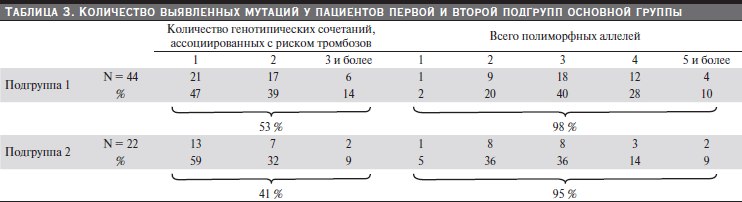
соответственно. Количество выявленных мутаций у пациентов первой и второй подгрупп основной группы представлено в табл. 3.
Статистически значимых различий по распределению полиморфизмов генов среди детей соматически здоровых и с тромботическим эпизодом в анамнезе не выявлено. Генотипические сочетания, ассоциированные с риском тромбозов (в табл. 2 выделены жирным шрифтом), отмечены в обеих подгруппах: у 53 % детей – в 1-й подгруппе, у 41 % – во 2-й. Чаще встречалось одновременное носительство мутантных аллелей генов MTHFR, PAI-1 и FGB.
Показатели клинических и биохимических анализов крови (общий белок, альбумин, креатинин, мочевина, мочевая кислота, холестерин, триглицериды, аланиновая и аспарагиновая трансаминазы, щелочная фосфатаза, электролитный состав), коагулограммы (фибриноген, активированное частичное тромбопластиновое время [АЧТВ], международное нормализованное отношение [МНО], тромбиновое время) соответствовали норме у всех детей (табл. 4). При сравнении групп по критерию
Манна–Уитни статистически значимые различия получены между 1-й и 2-й подгруппами в показателях тромбинового времени (p < 0,01).
СКФ рассчитана по формуле Шварца. У всех детей СКФ соответствовала возрастной норме и составляла: в 1-й подгруппе – 102,2 [IQR 97,5–109,9] мл/мин, во 2-й – 96,4 [IQR – 91,8–103,3], в группе контроля – 99,0 [IQR – 94,1– 108,4] мл/мин (p = 0,16 по критерию Краскела–Уоллиса).
При оценке клинического анализа мочи у детей 1-й подгруппы лейкоцитурия до 15 в п/зр зафиксирована у 2 девочек (при клиническом осмотре отмечены явления вульвита), эритроцитурия до 4–5 в п/зр – у 9 детей, во 2-й подгруппе у 7 детей также отмечена микроэритроцитурия до 5 в п/зр в
повторных анализах. Выявленные изменения нельзя считать статистически значимыми (р = 0,36 и р = 0,77 соответственно, двусторонний критерий Фишера). Микроальбуминурия в утренней порции мочи определялась у восьми пациентов 1-й подгруппы и стольких же пациентов 2-й, в группе контроля –
у семи детей, что также не является статистически значимым (р = 0,13 и р = 0,4 соответственно, двусторонний критерий Фишера).
Таблица 2. Распределение полиморфизмов ДНК у пациентов обеих подгрупп основной группы и здоровых лиц.
Таблица 3. Количество выявленных мутаций у пациентов первой и второй подгрупп основной группы.
По данным УЗИ, размеры почек соответствовали возрастным и расчетным показателям (в зависимости от массы тела ребенка). В среднем индекс почечной массы, определенный по стандартной методике (М.И. Пыков, 2001), в 1-й подгруппе был равен 0,42 ± 0,02 (M ± SD), во 2-й – 0,42 ± 0,03 (M ± SD), в группе контроля – 0,41 ± 0,03 (M ± SD). При сравнении результатов статистически значимых различий не получено (р = 0,35 критерий Стьюдента при сравнении 1-й и 2-й подгрупп, р = 0,36 при сравнении ANOVA всех трех групп). У одного ребенка раннего возраста из 1-й подгруппы выявлена умеренная пиелоэктазия слева до 7 мм и у двух детей группы контроля УЗ-признаки удвоения чашечно-лоханочной системы справа. При цветовом допплеровском картировании (ЦДК) определено хорошо
выраженное сосудистое дерево почки и наличие кровотока в мелких сосудах, который прослеживался в периферических отделах коркового слоя. При допплерографии сосудов почек изменений скоростных параметров кровотока и индекса резистентности (IR) не отмечено. IR на всех внутрипочечных сосудах каждого пациента были практически одинаковыми и составляли в 1-й подгруппе 0,67 [IQR – 0,65–0,68], во 2-й – 0,64 [IQR – 0,63–0,67], в группе контроля – 0,65 [IQR – 0,63–0,67]. Статистически значимые различия получены при сравнении 1-й и 2-й подгрупп (p = 0,01 критерий Манна–Уитни), незначимые – при сравнении подгрупп основной группы и контроля (р = 0,06; критерий Краскела–Уоллиса).
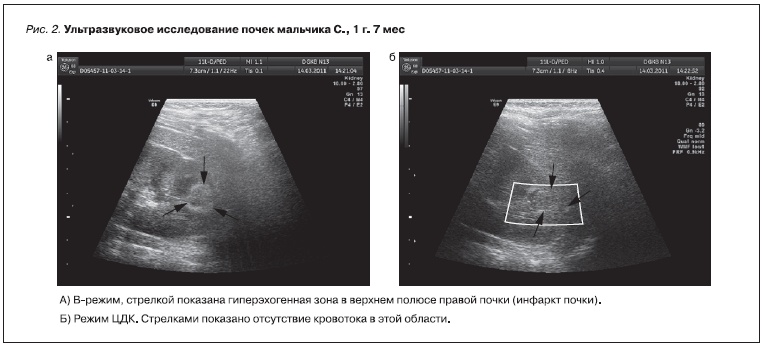
У семерых (пятеро детей 2-й подгруппы, двое – 1-й) с наследственной тромбофилией по данным УЗИ почек выявлены гиперэхогенные зоны треугольной формы в паренхиме почек с отсутствием кровотока в них, что было расценено как ИП (рис. 2 и 3). У пятерых детей с ультразвуковыми признаками ИП в анамнезе имелось указание на перенесенный тромбоз (дети второй группы), одна девочка обследована в связи с отягощенным семейным анамнезом (у мамы хронический гломерулонефрит и наследственная тромбофилия), один мальчик наблюдался в связи с сопутствующей патологией почек
(агенезия левой почки). Диагноз ИП не мог быть подтвержден другими, не УЗ, методами в связи с возможным риском тромбообразования у детей с наследственной тромбофилией при манипуляциях на сосудах и при введении контрастного вещества. В среднем от момента тромбоза до выявления ИП
прошло 18,2 ± 10,5 месяцев (M ± SD). Средний возраст на момент установления диагноза составил 7,6 ± 6,6 года (M ± SD). Из анамнеза троих детей (10, 16 и 3,5 года) известно, что ранее у них фиксировалась микроэритроцитурия до 5–7 в п/зр, которая рассматривалась как результат кристаллурии. Эпизодов повышения артериального давления не наблюдалось ни у одного ребенка. У двух детей (16 и 1,5 лет) при эпизоде острого тромбоза другой локализации определялись интенсивные жалобы на боли в правом боку, которые трактовались как функциональные нарушения ЖКТ в одном случае и подозрение на хирургическую патологию в другом (у девочки К. фиксирована киста правого яичника, однако перекрута, некроза и других изменений не диагностировано).
У детей с ИП по сравнению с детьми с наследственной тромбофилией, но без ИП чаще выявляли эритроцитурию (4/7 vs 12/59, р = 0,05), микроальбуминурию (5/7 vs 12/59, p = 0,01), никтурию (4/7 vs 8/59, р = 0,017), нарушения ацидо-аммониогенеза (5/7 vs 9/59, p = 0,0036). Уровень значимости р
рассчитан по двустороннему критерию Фишера.
Рисунок 2. Ультразвуковое исследование почек мальчика С., 1 г. 7 мес.
При исследовании ферментов мочи (арилсульфатаза-а, β-глюкуронидаза, γ-глутамилтрансфераза, холинэстераза) определено статистически значимое (двусторонний критерий Фишера, p < 0,001 во всех случаях) повышение лизосомальных ферментов (β-глюкуронидазы и арилсульфатазы-а), а также
γ-глутамил-трансферазы: в среднем до 0,67 [IQR – 0,65–0,9] 0,65 [IQR – 0,53–0,8] и 39,4 [IQR – 34,5–43,5] соответственно при норме до 0,45, 0,4 и 45 E/мг/мл креатинина соответственно, что свидетельствует о дисфункции канальцевого аппарата почки.
По данным коагулограммы на момент обследования признаков гиперкоагуляции не зафиксировано ни одного ребенка с ИП. При исследовании уровней естественных антикоагулянтов у четверых выявлено снижение протеина С, у одного – протеина S; уровень антитромбина III был нормальным у всех детей с ИП. Таким образом, вероятность развития ИП у детей с наследственной тромбофилией при наличии сниженного протеина С возрастает в 38 раз по сравнению с детьми, имеющими другие маркеры наследственной тромбофилии, но нормальный уровень протеина С (OR = 38 95 % CI: 3,6–549,3; RR = 13,3 CI: 2,9–39,3; p = 0,0007, двусторонний критерий Фишера).
При проведении генетических исследований выявлены полиморфизмы генов PAI-1 (-675 4G/4G) у шести детей, FV Leiden (1691 G/A) у одной девочки, фибриногена (-455 G/A) у двух детей, а также отмечено наличие гетерозиготного полиморфизма в гене MTHFR (у 4 детей), не относящегося к факторам, повышающим риск тромбозов при нормальном уровне гомоцистеина. При сравнении групп детей с участком ИП и детей без изменений в почках выявлены статистически значимые различия в присутствии гомозиготного полиморфизма в гене PAI-1: при наличии данного полиморфизма риск
развития ИП повышается в 10 раз (р = 0,016 двусторонний точный критерий Фишера, OR = 10,8, CI – 1,2–260,8; RR = 8,6, CI – 1,1–193,9).
К специальным биохимическим маркерам, исследованным в ходе данной работы, отнесено определение уровней липопротеина (а) (референсные значения – 0–30 мг/дл) и гомоцистеина (референсные значения – 3,7–13,9 мкмоль/л), при повышении которых увеличивается риск возникновения тромботических эпизодов.
В многочисленных работах [57–61] показано, что апо (а), находящийся в составе Лп (а), по строению сходен с плазминогеном и конкурирует с ним за места связывания как на молекуле фибрина, так и на эндотелиальных клетках, тем самым вытесняя плазминоген из мест его связывания. Таким образом, уменьшается образование плазмина, что в свою очередь снижает активность фибринолиза и способствует усилению тромбогенеза. Гомоцистеин обладает выраженным токсическим действием на эндотелиальную клетку. При избытке гомоцистеина в организме он начинает накапливаться в крови
и основным местом повреждения становится внутренняя поверхность сосудов.
Уровень липопротеина (а) у детей с ИП был значимо выше по сравнению с детьми с наследственной тромбофилией: 83,0 [IQR – 59,7–175,5] и 7,7 [IQR – 4,7–14,1] соответственно (p = 0,00037, критерий Манна–Уитни). У девочки 3,7 года с ИП, сниженным уровнем протеина С, Лейденской мутацией
и полиморфизмом 4G/4G гена PAI-1 уровень липопротеина (а) превышал нормативные значения лишь на 2,6 мг/дл, у остальных 6 детей липопротеин (а) был выше в 2–9 раз. При исследовании липопротеина (а) у детей с наследственной тромбофилией, но без ультразвуковых признаков ИП лишь
у одной девушки 17,3 года зафиксировано повышение липопротеина (а) до 111 мг/дл, у остальных детей уровень липопротеина (а) оставался нормальным. Таким образом, повышение липопротеина более чем в 2 раза у детей с наследственной тромбофилией соответствует увеличению риска более чем в 300 раз (р < 00001 по двустороннему критерию Фишера, OR = 348, 95 % CI – 14,4–51554; RR =
50,6, CI: 7,9–655,5), чувствительность данного метода составляет 84 % (CI – 48,5–98,7), специфичность – 98 % (CI – 93,9–99,9).
Средний уровень гомоцистеина у детей с ИП был равен 7,86 (IQR – 7,2–12,0), у детей с наследственной тромбофилией без ИП – 7,9 (IQR – 6,14–10,7) по критерию Манна–Уитни, р = 0,6.
Уровень гомоцистеина также не различался между детьми 1-й и 2-й подгрупп основной группы (8,85 (IQR – 6,0–11,0);
7,55 (IQR – 6,74–9,64) р = 0,53, критерий Манна–Уитни), но оказался выше по сравнению с детьми контрольной группы (в контрольной группе средний уровень гомоцистеина был равен 5,3 (IQR – 4,5–6,5), р = 0,007, критерий Краскела–Уоллиса). Умеренная гипергомоцистеинемия в сочетании с
полиморфизмом в гене MTHFR выявлена у 7 детей: у 3 – из 1-й подгруппы и у 4 – из 2-й. Необходимо отметить, что у 10 детей с гомозиготной мутацией в гене MTHFR уровень гомоцистеина оставался нормальным, причем отчетливо прослеживалась связь с возрастом ребенка: при наличии одинаковых
полиморфизмов гена у детей старшего возраста по сравнению с младшими уровень гомоцистеина был выше (коэффициент корреляции Спирмена Rs = 0,69, n = 45; р < 0,0001). Однако связи изменения функционального состояния почек у детей с повышением уровня гомоцистеина в крови не выявлено.
Рисунок 3. Ультразвуковое исследование почек мальчика А., 2 года.
При выявлении ИП всем детям по согласованию с врачом-гематологом назначена антикоагулянтая терапия, если пациент не получал ее раньше. Для улучшения клеточного энергообмена все дети получали антигипоксанты и антиоксиданты (цитохром С, коэнзим Q10, свечи корилип®, рибоксин® и другие препараты по стандартным схемам). За время катамнеза (7–28 месяцев) новых эпизодов тромбоза сосудистого русла почек выявлено не было. Самочувствие детей, перенесших ИП, оставалось удовлетворительным.
Таким образом, дети с наследственной тромбофилией, у которых развился инфаркт участка почки, требуют диспансерного наблюдения не только у гематолога, но и у нефролога, т. к. могут быть отнесены к группе пациентов с хронической болезнью почек (ХБП). ХБП по определению KDOQI (2002) – это повреждение почек независимо от его природы, которое прослеживается 3 месяца и более и проявляется нарушением структуры и/или функции почек.
К первой стадии ХБП могут быть отнесены 6 детей с ИП в связи с СКФ > 90 мл/мин, наличием микроальбуминурии, минимального мочевого синдрома (микроэритроцитурия), ко второй степени – 1 мальчик (СКФ = 86 мл/мин, наличие микроальбуминурии).
С учетом минимальных клинических проявлений, развития тяжелых осложнений и возможной инвалидизации больных была разработана схема обследования детей с наследственной тромбофилией. Оценка риска развития тромбоза сосудов почек у детей и тактики ведения пациентов при его выявлении основывается на выделении групп низкого и высокого рисков по развитию тромбоза артериальных сосудов почки у детей на основании определения уровня липопротеина (а), активности
естественных антикоагулянтов, в первую очередь протеина С, а также результатов УЗИ и УЗДГ сосудов почек. В группу риска по развитию ИП могут быть отнесены пациенты с высоким уровнем липопротеина (а), превышающим более чем в 2 раза контрольные значения. Помимо наблюдения гематолога дети с высоким риском развития ИП должны наблюдаться у нефролога с включением в терапию не только антикоагулянтов, но и препаратов, улучшающих клеточный обмен и микроциркуляцию (антигипоксанты, антиоксиданты).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}