
Меняется ситуация в мире, все большее значение приобретает действие экологических влияний, роль которых не может не сказаться на генетическом аппарате всех живых существ, что ведет к необходимости выявлять заболевания, связанные с мутацией различных генов. Сегодня особенно
актуальным стало представление о том, что многие хронические болезни взрослого человека формируются в детстве начиная с внутриутробного периода [1].
Одна из важных проблем нефрологии – ранняя диагностика наследственных нефропатий и структурного дизэмбриогенеза почек [2, 3]. Данная проблема актуальна для любого возрастного периода, т. к. первым проявлением своевременно не выявленных в детском возрасте врожденных аномалий органов мочевой системы (ОМС) у взрослых нередко может быть снижение почечных функций [4].
Нарушение антенатального развития почек нередко сочетается с пороками развития нижних мочевых путей. Термин CAKUT (congenital anomalies of kidney and urinary tract) – наличие врожденных анатомических аномалий почек и мочевого тракта, появился сравнительно недавно – фактически когда на ранних сроках беременности при использовании УЗИ начали обращать внимание на развитие органов мочевой системы. Однако о возможности одновременного развития врожденных аномалий почек и нижних мочевых путей говорилось в руководствах А.Я. Пытеля и С.Д. Голигорского уже в середине прошлого столетия [5], причем рентгенологическое описание врожденных болезней почек и мочевого тракта дано А.Я. Пытелем и Ю.А. Пытелем уже в 1965 г. [6].
Известно, что почки и мочеточники развиваются из мезодермы, а мочевой пузырь и уретра имеют энтодермальное происхождение и возникают из урогенитального синуса. До формирования окончательной почки имеется две промежуточные почки – пронефрос и мезонефрос. Пронефрос
нефункционален, но его проток становится протоком мезонефроса, который вслед за этим развивается в мочеточниковый зародыш. К 6-й неделе внутриутробного развития мочеточниковый зародыш и метанефрогенная бластема являются двумя компонентами, поддерживающими образование окончательной почки – метанефроса [7].
Одновременно с развитием почки происходит и формирование мочевых путей. Параллельно с образованием мочеточникового зародыша от клоаки отшнуровывается урогенитальный синус и сливается с протоком мезанефроса. С 6-й по 34-ю неделю гестационного периода продолжается
процесс нефрогенеза. После 36-й недели гестации почка имеет полный набор нефронов.
В мировой литературе в настоящее время наиболее часто при CAKUT приводятся данные о его частоте в различных популяциях и результаты поиска генов, мутация которых может вызывать этот синдром. В Европе создан консенсус по поводу возможности включения в работу многих центров различных европейских стран для ранней диагностики и лечения этого тяжелого заболевания [8, 9]. Ищут различные пути, чтобы подобраться к пониманию развития этого тяжелого порока [10]. Серьезные исследования проводятся в экспериментах на животных [11]. Разрабатываются методики прогнозирования CAKUT у детей [12, 13].
CAKUT – это одно из наиболее опасных состояний у новорожденных, нередко приводящих к летальному исходу [14, 15, 16]. Если в антенатальном периоде при УЗИ определен CAKUT-синдром, родителям предлагают решить вопрос о преждевременном прерывании беременности. CAKUТ-
синдром может выявляться у подростков и даже у взрослых людей [17]. Нет единых данных о частоте CAKUT как в антенатальном периоде развития ребенка, так и в различные возрастные периоды после рождения. В 2003 г. сообщалось, что частота CAKUT-синдрома в Японии составляет 1 случай на 500 новорожденных [18]. Развитие порока происходит в эмбриональном периоде – этапе формирования почек и мочевыделительной системы [19]. При наблюдении за развитием 822 детей в Швейцарии, Германии, Турции и Камеруне, у которых в антенатальном периоде определен CAKUT-синдром, было показано, что наиболее тяжело протекает заболевание, если выражен гидронефроз [20]. По-видимому, оптимально одновременное наблюдение таких больных нефрологами и урологами [21].
При CAKUT-синдроме наиболее часто встречается присоединение рецидивирующей инфекции мочевой системы (ИМС), обычно принимающей характер рецидивирующего пиелонефрита, несколько реже развивается артериальная гипертензия [22].
При ультразвуковом скринировании в антенательном периоде 26 989 детей Китая у 484 (1,67 %) заподозрен CAKUT-синдром. При рождении большинство из них не имели клинических проявлений болезни. Исключение составляли дети (преимущественно мальчики) с гидронефрозом [23]. Во всех
случаях УЗ-скринирование позволяет лишь предположить CAKUT. Для уточнения диагноза во всех случаях необходимо использование рентгеноурологических методов исследования.
В США, по данным I.V. Vosypin [24], CAKUT-синдром встречается у 3–6 из 1000 живорожденных новорожденных, причем это оказывается предрасполагающим фактором к раннему развитию артериальной гипертензии и других вариантов поражения сердечно-сосудистой системы.
Экспериментальные данные говорят, по мнению I.V. Vosypin, не об одной, а о нескольких причинах развития этого синдрома. К ним относятся генетические мутации, эпигеномные воздействия, а также внешние экологически вредные влияния. Специальные исследования проведены в США по определению роли группы ферментов (гистон диацелатоз (HDACs), обладающих эпигеномным влиянием на развитие почек и мочевого тракта в эксперименте на мышах [25].
По данным 12 стран Европы при УЗИ 700 тыс. детей в антенатальном периоде CAKUT был выявлен в 0,008 % случаев [26]. Близкие показатели у живорожденных новорожденных получены в Японии [27].
Частота CAKUT выше в тех семьях, где у родственников имеются случаи аномалий органов мочевой системы. В настоящее время сообщается, что более 500 синдромов могут быть с выраженным CAKUT [10]. По мнению S. Weber [10], в 10 % случаев CAKUT-синдром имеет генетическое происхождение. К этому мнению присоединяются многие нефрологи и генетики [28, 29]. Возможность генетического происхождения начали искать в конце ХХ и начале ХХI вв. Были исследованы 52 гена – кандидата развития CAKUT [30]. Наибольшее внимание было обращено на те случаи CAKUT, которые имели
проявления нефропатии, развившейся в связи с тяжелым пузырно-мочеточниковым рефлюксом [31, 32, 33]. В случаях пузырно-мочеточникового рефлюкса анализировали значимость 44 генов – кандидатов развития CAKUT-синдрома. К ним были отнесены гены группы SNP3, ROBO2, EYA1, CREM1, UPS3A. M. Hiraoka et al. [27] провели серьезные исследования значимости мутации гена AT2R как причины развития CAKUT. Однако сравнение с популяционной частотой этой генетической аномалии заставило авторов отказаться от своей гипотезы, по крайней мере по отношению к японской популяции.
В антенатальном периоде развития как у экспериментальных животных (в основном мыши), так и у человека большая роль принадлежит гену PAX2 [34, 35]. По мере изучения роли Pax2 в развитии мочевой системы оказалось, что отрицательное влияние на нефрогенез и органы мочевыведения оказывает взаимоотношение PAX2 с такими транскрипционными факторами, как Gdnt, Ret, SHH, Wnt 4, Fgt [36]. Специальное внимание M. Paces-Ferry et al. [37] обращают на влияние фактора Hnf-1b на PAX2, который в эксперименте на мышах усиливает влияние PAX2-гена на развитие почки.
Гетерозиготная мутация этих генов у человека ведет к развитию аномалий различных отделов мочевого тракта, влияет на их морфогенез.
Сделаны попытки выделить специфические гены, мутация которых приводит к развитию CAKUT-синдрома [38]. Проверены больные как в антенатальном, так и в постнатальном периодах. Оказалось, что возможно развитие патологии при наличии мутации одного гена, но может наблюдаться
комбинация мутирующих генов. Авторы проверили наличие мутации FRAS1 и EREM2 при развитии CAKUT. Сравнение осуществлено со здоровыми, у которых не было врожденной патологии почек и органов мочевыведения и подобной мутации. В исследованиях E. Pavlakis et al. [39] указано на роль мутации генов FRAS1/FREM, ответственных за состояние гломерулярных базальных мембран на самых
ранних периодах развития эмбриона, что может проявиться при CAKUT.
H. Cordell et al. [40] проверили возможность быть причиной CAKUT гены ПМР как одного из его проявлений – AGTR2, HNF1, PAX2, RET и UPK34. Результаты оказались отрицательными. Поставлен вопрос о необходимости дальнейших исследований. A. Brockschmidt et al. [41] предлагают новый
кандидат в мутирующие гены при CAKUT – CHD1L. Однако, по-видимому, наиболее реальными причинами развития врожденной аномалии почек и мочевых путей является одновременная мутация PAX2 и EMX2 [42]. Сочетания подобной мутации не обнаружено у здоровых эмбрионов как в эксперименте на мышах, так и у человека, причем у людей оба гена находятся на 10q-хромосоме, их мутация сопровождается полной деструкцией хромосомы.
При исследованиях причин развития CAKUT-синдрома в работах последнего времени обращается внимание на роль первичных цилий в развитии патологии. По-видимому, пришло время изучать мутации генов, кодирующих белки аксонемы первичных цилий [43, 44]. Возможно, именно гены,
кодирующие белки первичных цилий, помогут разобраться в причинах развития как изолированного, так и синдромального CAKUT.
C целью характеристики синдрома CAKUT у представителей отечественной популяции все дети (в течение года 500 больных), находившихся на лечении в нефрологическом отделении ФГБУ МНИИПиДХ Минздрава России обследовались общеклинически, включая определение функционального состояния почек. При наличии у ребенка ИМС и/или пузырно-мочеточникового рефлюкса (ПМР) исследование и наблюдение проводились нефрологом совместно с урологом. Обращалось внимание на состояние различных органов, в которых могли быть признаки дизэмбриогенеза, в т. ч. подсчитывалось количество малых аномалий развития (МАР) – стигм соединительнотканного нарушения, связанных с органным изменением эмбриогенеза. Специально проанализировали характер CAKUT на рентгенограммах 42 детей и сопоставили с данными УЗ-скринирования, позволившими предполагать наличие этого синдрома. Основной акцент был сделан на определении состояния органов мочевой
системы на основании УЗ-исследования. УЗ-скринирование осуществлялось на аппарате Voluson – 730 Expert, GE (США). При подозрении на наличие врожденных аномалий ОМС проводилась внутривенная экскреторная урография и/или микционная цистография с использованием аппаратов Duo Diagnost 2004, Philips (Голландия). В качестве контрастного вещества вводился Йогексол (Омнипак) в количестве, соответствующем возрасту ребенка и функциональному состоянию почек. Отдельно были выделены группы детей с наследственным нефритом (синдром Альпорта) и АДПБП.
Хорошо известно, что в синдром CAKUT включаются различные аномалии почек и органов мочевыведения: это могут быть агенезия почки, гипо- и дисплазия почечной ткани, удвоение мочеточников, изменения в мочевом пузыре, клапаны уретры и др. пороки [8]. Ряд из этих аномалий мы
выявили при УЗ-скринировании и рентгено-урологическом обследовании. Когда выявляются только аномалии органов мочевой системы, говорится об изолированном CAKUT. Однако очень редко дизэмбриогенез органов мочевой системы существует изолированно. Как правило, имеет место и нарушение развития других органов, прежде всего при осмотре выявляется множество внешних признаков соединительнотканного дизэмбриогенеза. При специальном обследовании выявляются и другие аномалии. В таких случаях обычно используется термин “синдромальный CAKUT”.
Клинический пример 1
Арина Р., русская (обследована в МНИИПиДХ в 2012 г.). Девочка от III беременности, протекавшей неблагоприятно – со склонностью к прерыванию, от I родов кесаревым сечением. Масса тела при рождении – 2600 г. Оценка по шкале Апгар – 6/8 баллов. У ребенка имела место внутриутробная пневмония, в течение двух недель девочка пребывала на искусственной вентиляции легких. Наблюдается пульмонологом по поводу бронхиальной астмы. В возрасте 3 месяцев были явления, расцененные как острый пиелонефрит, получала курс антибактериальной терапии с эффектом, однако ИМС рецидивировала. По данным в/в экскреторной урографии, проведенной в возрасте 4 лет, у девочки имело место удвоение ЧЛС с обеих сторон. При поступлении в МНИИПиДХ АД – 90/60 мм рт. ст. При осмотре отмечено 7 МАР. При обследовании выявлены признаки тяжело протекающей бронхиальной астмы с явлениями бронхо-легочной дисплазии, резко повышены IgE крови. При УЗИ ОМС выявлено увеличение объема почек, удвоение чашечно-лоханочной системы (ЧЛС) обеих почек, стенки собирательной системы почек утолщены. Определяется взвесь в просвете мочевого пузыря. При экскреторной урографии выявлено удвоение ЧЛС (рис. 1) и умеренное повышение подвижности левой почки. При микционной цистографии определялись признаки нейрогенной
дисфункции мочевого пузыря.
Рисунок 1. Клинический пример 1.
Электрокардиограмма (ЭКГ): миграция водителя ритма, резко выражена аритмия с периодами брадикардии. Косвенные признаки неполной блокады правой ножки Гиса.
Заключение: у девочки с тяжелым течением антенатального периода выявлены множественные признаки дизэмбриогенеза: 7 МАР, патология со стороны сердца, легких, иммунной системы с развитием тяжелой бронхиальной астмы и явления CAKUT, осложненные рецидивирующей ИМС.
Одним из наиболее тяжелых проявлений CAKUT является ПМР, который нередко оказывается основным вариантом врожденной аномалии почек и мочевыводящего тракта [40].
Клинический пример 2
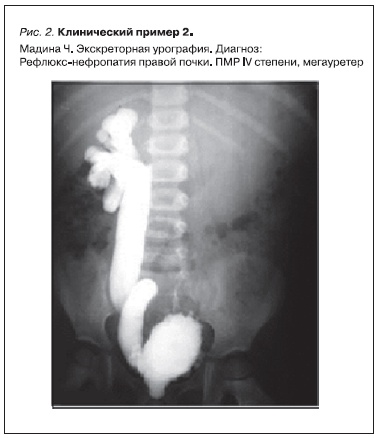
Мадина Ч. из Чеченской Республики 15 лет, наблюдается в МНИИПиДХ с 4-летнего возраста в связи с аномалиями ОМС. Девочка из семьи, где у двух младших сестер имеется аномалия ОМС, патология почечных сосудов у матери. Мадине с 7-месячного возраста поставлен диагноз: рецидивирующий
обструктивный пиелонефрит. В возрасте 4 лет в отделении нефрологии МНИИПиДХ при осмотре обращено внимание на множество МАР. На экскреторных урограммах выявлены гипоплазия и рефлюкс-нефропатия правой почки (рис. 2), дольчатость левой почки. При микционной цистографии
определен правосторонний ПМР III степени, т. е. признаки нейрогенной дисфункции мочевого пузыря. На динамической реносцинтиграфии, проведенной в то же время, подтверждена гипоплазия правой почки, а также обнаружен ПМР, отмечено выраженное нарушение выделительной функции обеих почек за счет задержки эвакуации из паренхимы и преходящей задержки эвакуации из собирательной системы. На статической реносцинтиграфии – диффузно-очаговые изменения обеих почек. Общий объем функционирующей паренхимы снижен.
Рисунок 2. Клинический пример 2.
При микционной цистографии отмечен нейрогенный мочевой пузырь, ПМР справа IV степени. На цистоскопии выявлены явления кистозного цистита; дистопия устья правого мочеточника; меатостеноз.
В 11 лет впервые выявлена артериальная гипертензия до 170/100 мм рт. ст. Повышение АД было медикаментозно скорригировано. Однако в дальнейшем антигипертензивная терапия проводилась нерегулярно, артериальная гипертензия сохранялась. В 11 лет у девочки впервые отмечены эпизоды
креатининемии. С 15 лет креатининемия (> 120 мкмоль/л) постоянная.
Офтальмолог выявил астигматизм. ЭКГ: синдром ранней реполяризации желудочков.
Диагноз. Рефлюкс-нефропатия. Вторичный обструктивный пиелонефрит, рецидивирующее течение. ХБП II стадии (СКФ – 67 мл/мин/1,7 м2). Нейрогенная дисфункция мочевого пузыря. Ренальная артериальная гипертензия. Синдром ранней реполяризации желудочков сердца. Миопия слабой степени. Миопический астигматизм. Ангиопатия сетчатки OU.
Заключение: у девочки из семьи, где у близких родственников выявлены различные заболевания почек врожденного характера, выражена рефлюкс-нефропатия. Ребенок страдал и другими врожденными аномалиями, которые, по-видимому, развились одновременно с CAKUT в эмбриональном периоде.
Изолированное наличие у больного ПМР встречается очень редко, обычно ПМР сочетается с дизэмбриогенезом других отделов мочевой системы. Нередко развивается синдромальное поражение, включающее глаза, нервную систему и различные паренхиматозные органы [45], что и наблюдалось у обоих описанных больных.
Нами был проведен анализ частоты CAKUT – синдрома при некоторых наследственных заболеваниях (см. таблицу). У 12 из 22 детей с синдромом Альпорта при УЗ-исследовании были заподозрены аномалии ОМС: ПМР (2/22), удвоение ЧЛС (2/22), дистопия и ротация почек (2/22); кроме того, у 27,3 % (6/22) пациентов выявлена пиелоэктазия, у 22,7 % (5/22) – признаки нейрогенной дисфункции мочевого
пузыря (НДМП). Им проведено рентгеноурологическое обследование: всем 12 детям – внутривенная урография и 11 пациентам (с пиелоэктазией и/или признаками НДМП) – микционная цистография. При внутривенной урографии наличие врожденных аномалий ОМС подтверждено в 50 % случаев (у 6/12), из них по 1 случаю удвоения ЧЛС и дистопии почек, у 2 из 6 детей с пиелоэктазией по результатам УЗИ выявлена обструкция мочеточников, у 1 – синдром Фрейли, у 1 – гидронефроз. При цистографии ПМР выявлен у 18,2 % (2/11) пациентов. Таким образом, в отношении 8 из 12 детей с подозрением на CACUT при УЗ-скринировании он подтвержден рентгеноурологически (см. таблицу).
Таблица. Патология мочевых путей у детей с синдромом Альпорта (данные рентгеноурологического обследования)
Наиболее тяжелым оказалось сочетание наследственного нефрита и гидронефроза.
Клинический пример 3
Коля К. 1994 г. рождения из семьи, где у его матери в 3 года выявлена гематурия. К моменту рождения мальчика матери поставлен диагноз наследственного нефрита с явлениями ХПН, проведен гемодиализ. У Коли в 1 год 8 месяцев выявлена гематурия. В 4 года мальчику поставлен диагноз “гидронефроз” (рис. 3), по поводу которого в 5-летнем возрасте проведена операция. При биопсии почки в возрасте 6 лет при электронной микроскопии найдены изменения, укладывающиеся в морфологическую картину наследственного нефрита (рис. 4).
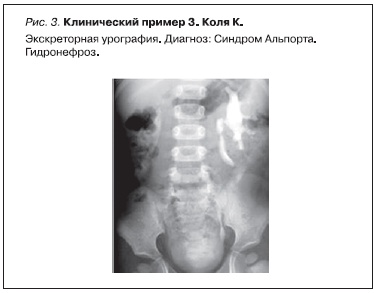
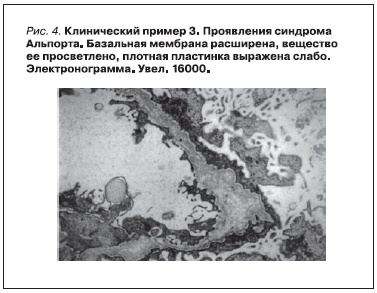
Заключение: у мальчика от матери, страдавшей наследственным нефритом, родился ребенок, у которого на 2-м году жизни обнаружена гематурия, при электронной микроскопии нефробиоптата в 6 лет выявлена типичная картина СА. Наличие гидронефроза, устраненного оперативным путем, предполагает, что CAKUT явился одним из проявлений патологии.
При обследовании 37 детей с АДПБП в нефрологическом отделении МНИИПиДХ CAKUT на основании УЗИ почек заподозрен у 8 (21,6 %) больных: выявлено увеличение лоханок; кроме того, у одного ребенка выявлен нефроптоз и у одного при осмотре – гипоспадия. По данным литературы, изучение состояния более 800 детей из Турции, Камеруна, Швейцарии и Германии c определением роли нефроцистина 3 как показателя развития медуллярного кистоза почек выявило у детей одновременно с кистозными изменениями в почках врожденные аномалии паренхиматозных органов и CAKUT-синдром [46].
Таким образом, при обследовании детей в нефрологическом отделении ФГБУ МНИИПиДХ Минздрава России встречался, как правило, синдромальный вариант CAKUT с явлениями дизэмбриогенеза других органов (множество МАР, аномалии проводящей системы сердца, бронхолегочной дисплазии и др.). Об этом говорят и некоторые литературные данные [20, 23]. При наличии у больного ИМС, особенно рецидивирующего пиелонефрита, а также в случае наличия у ребенка ПМР обследование и лечение
осуществлялись совместно нефрологом и урологом. О том, что подобный тандем несомненно полезен, показало комплексное обследование и лечение, которое было проведено Г.А. Маковецкой и соавт. [21]. В работе представлены данные о 265 детях в возрасте от месяца до 17 лет с врожденным
гидронефрозом и мегауретером, наблюдаемых с 1999 по 2010 г. У 145 из них ретроспективно проанализированы исходы патологии. Проспективные наблюдения выполнены 120 детям раннего возраста также с гидронефрозом, мегауретером. Своевременное урологическое вмешательство уменьшило число случаев развития ХПН (рис 5).
![Отдаленные результаты лечения детей с мегауретером (%, %) (Г. А. Маковецкая и др. 2011 год [21])](https://lib.medvestnik.ru/apps/lib/assets/uploads/nephrology/2013/2/neph-2-2013-pic-38.jpg)
По-видимому, УЗ-скрининг позволяет заподозрить наличие CAKUT. Однако окончательное заключение может быть дано только после рентгеноурологического обследования. Для уточнения данного положения проведено сравнение результатов УЗИ, экскреторной урографии и/или микционной цистографии 42 детей, у которых, как правило, была рецидивирующая инфекция мочевых путей. Вероятно, можно разделить показания к экскреторной урографии по УЗ-признаку на две группы: абсолютные и относительные. К абсолютным стоит отнести аномалии формы, расположения,
количества почек, кисты почек, пиелоэктазию, которые могут быть обнаружены при УЗИ. Поэтому вряд ли стоит отказываться от экскреторной урографии при аутосомнодоминантной поликистозной болезни почек, т. к. при УЗИ выявляются далеко не все проявления врожденной аномалии мочевыводящей системы.
К относительным показаниям стоит отнести все остальные УЗ-признаки, включающие изменения паренхимы почек, утолщение стенок собирательной системы почек, утолщение стенок лоханок в совокупности с клинической картиной заболевания. В урологической клинике внутривенная урография
является обязательной диагностической манипуляцией при подозрении на наличие мочекаменной болезни, и CACUT при этом также нередко становится диагностической находкой [47]. Урологи предлагают специально готовить к урографическому обследованию мочевой системы [48]. Это положение безусловно относится и к больным, которым экскреторная урография проводится после УЗ-скринирования.
В качестве краткого заключения следует повторить, что УЗ-скрининг ОМС у детей дает возможность лишь заподозрить наличие CAKUT, поэтому при подозрении на врожденные аномалии ОМС целесообразно проводить рентгеноурологическое обследование. Кроме этого следует обращать внимание на проявления дизэмбриогенеза со стороны других органов.
Термин “CAKUT-синдром”, отражающий наличие врожденных аномалий почек и/или мочевого тракта, в
настоящее время стал общепринятым. Наиболее полноценное клиническое изучение CAKUT должно проводиться одновременно нефрологами и урологами. Нередко кроме CAKUT выявляется дизэмбриогенез других органов. По данным литературы, 10 % CAKUT генетической природы. Проверен ряд кандидатов в гены, в настоящее время считается, что наиболее вероятная причина CAKUT лежит в одновременной мутации PAX2- и EMX2-генов. Имеет значение эпигеномное влияние и воздействие внешних тератогенных факторов. Важно выявлять CAKUT в антенатальном периоде и периоде новорожденности при использовании УЗ-скринирования. При позднем выявлении этой патологии у детей старшего возраста и у взрослых нередко определяется уже формирование почечной недостаточности. Имеются данные, говорящие о наличии CAKUT при наследственной патологии, где основное внимание уделено только состоянию почки. Наибольшее внимание среди проявлений CAKUT привлекают тяжелые формы ПМР, следствием которого нередко оказываются рефлюкс-нефропатия и гидронефроз. При CAKUT часто развивается рецидивирующий пиелонефрит. По-видимому, необходимость уточнения CAKUT требует более широкого использования для диагностики у детей и взрослых не только УЗИ, но и рентгеноурологических методов исследования с применением современных малотоксичных препаратов.



{kind=link}
{kind=link}
{kind=link}