
Введение
Гематурия — один из наиболее частых симптомов, характерных для поражения почек и мочевых путей [1—5]. Под гематурией (ГУ) понимают наличие ≥5 эритроцитов в поле зрения при х40-микроскопии мочи, центрифугированной при 750 g, либо более 3 эритроцитов в поле зрения нецентрифугированной мочи, более 1 млн эритроцитов в 24-часовой моче (проба Аддиса) [2, 3, 5]. ГУ, определяемую только при микроскопии осадка мочи, называют микрогематурией. В случае если количество присутствующих в моче эритроцитов приводит к изменению цвета мочи, что становится видно невооруженным глазом (“цвет мясных помоев/кока-колы”), такое состояние называется макрогематурией. Изменение цвета мочи достигается очень небольшим содержанием крови: 0,5—1,0 мл на 1 л мочи [2, 3, 5]. Микрогематурия может часто протекать бессимптомно, не обращать на себя внимания долгие годы и обнаружиться случайно при рутинном обследовании пациента, тогда как может являться признаком серьезных заболеваний, ведущих к снижению почечных функций вплоть до терминальной почечной недостаточности. Микрогематурия может также сочетаться с клиническими симптомами (дизурия, лихорадка, боли, повышение артериального давления, геморрагичексий синдром и т. д.), проявляться изолированно либо сопровождаться протеинурией [2, 6, 7].
Принципиальное значение в плане дальнейшей тактики обследования и ведения пациента имеет определение характера гематурии. На ренальный характер указывает сочетание ГУ с протеинурией и/или недостаточностью почечных функций. Однако нередко гематурия является единственным симптомом. В этом случае скрининговым методом для разделения гематурии гломерулярного и негломерулярного характера можно считать выявление морфологически измененных (дисморфных) форм эритроцитов (изменение формы, размера, содержание гемоглобина) при помощи фазово-контрастной микроскопии [2, 8—10]. Наибольшее значение имеет обнаружение особых форм дисморфных эритроцитов — акантацитов, которые характеризуются “прыщевидными” выпячиваниями клеточной стенки. Обнаружение в моче 5 % и более акантацитов, а также эритроцитарных цилиндров, которые представляют собой эритроцитарные слепки гломерулярных канальцев, является достоверным маркером гломерулярной патологии [5, 8]. В случае подтверждения гломерулярного характера гематурии в большинстве случаев показано проведение нефробиопсии для выяснения причины, вызвавшей синдром ГУ.
Особенностью большинства гломерулопатий, проявляющихся гематурией у детей, является сходная клиническая и светооптическая морфологическая картина, что затрудняет дифференциальную диагностику между этими патологиями [11, 12]. В большинстве случаев в основе изолированной персистирующей дисморфной гематурии у детей и взрослых лежит три гломерулярных заболевания: синдром Альпорта (СА), болезнь тонких базальных мембран (БТБМ) — эти заболевания вызваны мутацией в гене COL4A3-5, что приводит к нарушению строения гломерулярной базальной мембраны, и IgA-нефропатия [13].
Помочь в постановке точного диагноза может проведение иммуногистохимического исследования цепей коллагена. Метод основан на формировании иммунных комплексов в результате присоединения специфичных антител, выделенных у животных (например, мышей), к антигенам соответствующей цепи. В случае отсутствия или измененного строения специфичные антитела не обнаруживают соответствующих антигенов, не присоединяются к ним и в результате — в пробе не получается окраски искомых объектов или окраска получается фрагментарной и бледной в случае патологического строения цепи. В рамках синдрома Альпорта и болезни тонких базальных мембран функциональность α3-, α4-, α5-тримеров коллагена IV типа всегда нарушается. Различные варианты СА характеризуются поражением различных цепей коллагена. При Х-сцепленном СА в основном поражается α5-цепь коллагена, которая является основной цепью сетей коллагена IV типа (α3, α4, α5 и α5, α5, α6), формирующих гломерулярную базальную мембрану. Аутосомные варианты СА, так же как и БТБМ, характеризуются поражением α3- и α4-цепей коллагена IV типа [14]. Поэтому дифференциация отдельных α-цепей коллагена IV типа посредством иммуногистохимии может быть очень ценным указанием [4, 15]. Определение этиологической структуры гломерулярной гематурии у детей — жителей Российской Федерации стало целью настоящего исследования.
Материал и методы
С целью выяснения причин гломерулярной ГУ в российской педиатрической популяции нами было проведено ретро- и проспективное обследование 57 пациентов нефрологического отделения Института педиатрии НЦЗД РАМН, страдающих гематурией: 14 детей с изолированной ГУ, 35 пациентов с ГУ в сочетании с протеинурией до 1 г/л, 2 ребенка с ГУ в сочетании с протеинурией более 1 г/л, но менее 3 г/л, 6 пациентов с ГУ в сочетании с протеинурией > 3 г/л, но без признаков нефротического синдрома в качестве первоначальных проявлений заболевания: 43 (75 %) мальчика, 14 (25 %) девочек, средний возраст — 13 ± 3 года. Средняя длительность болезни на момент проведения биопсии составила 5 ± 4 года. Пациентам с изолированной гематурией, в т. ч. макрогематурией и ГУ в сочетании с протеинурией следового уровня, до проведения биопсии был подтвержден гломерулярный характер ГУ при помощи фазово-контрастной микроскопии свежевыпущенной мочи.
Общий анализ клинико-лабораторных данных в динамике за 10 лет выявил сохранение изолированной ГУ у 10 (17,5 %) обследованных, сочетание ГУ с протеинурией до 1 г/л у 20 (35 %) детей, учащение ГУ в сочетании с протеинурией более 1 г/л, но менее 3 г/л до 10 (17,5 %) случаев, ГУ в сочетании с протеинурией 3 гл и более до 17 (30 %) случаев. Семейный характер гематурии выявлен у 27 (47%) обследованных, макрогематурия — у 23 (40 %) пациентов. Связь дебюта болезни с предшествовавшими инфекционными заболеваниями обнаружена у 21 (37 %) обследованных. У 18 (31,5 %) пациентов развилась нейросенсорная тугоухость, у 14 (24,5 %) — патология зрения. Для 16 (28 %) больных отмечено присоединение артериальной гипертензии. У 8 (14 %) обследованных произошло снижение скорости клубочковой фильтрации.
Всем детям была проведена нефробиопсия с последующим световым, иммуногистохимическим и электронным исследованиями нефробиоптата. Биопсия проводилась чрескожным доступом под прямым ультразвуковым контролем, изымалось по 2 столбика почечной ткани из нижнего полюса левой почки, при помощи биопсийной иглы под местной анестезией или под наркозом в зависимости от возраста пациента. Один из столбиков фиксировался в 10 %-ном растворе формалина и после изучался при помощи световой микроскопии (методы окраски: гематоксилин-эозин, пикрофуксин по ван Гизону, по Масону—Голднеру, ШИК-реакция). Второй столбик почечной ткани фиксировался в 2,5 %-ном растворе глютарового альдегида с последующим исследованием при помощи электронной микроскопии.
Результаты и обсуждение
Проведенное светооптическое исследование выявило, что наиболее частой морфологической находкой при синдроме ГУ является мезангиальная пролиферация 66 %, 19 % случаев сопровождались фокально-сегментарным гломерулосклерозом (ФСГС). У 10 (17 %) обследованных в интерстиции кортикального слоя определены пенистые клетки. После проведения электронной микроскопии было установлено следующее: наибольшее число пациентов страдало патологией COLIV — 37 (65 %) обследованных, из них у 20 (35 %) выявлен синдром Альпорта (СА), у 17 (30 %) — болезнь тонких базальных мембран (БТБМ), число пациентов, страдавших IgA-нефропатией, было сопоставимо с СА — 20 (35 %) обследованных.
Важно, что даже после проведения электронной микроскопии для 13 (35 %) пациентов с патологией COL4 дифференцировать СА и БТБМ не представлялось возможным в связи с неоднозначной морфологической картиной, которая характеризовалась в основном истончением гломерулярной базальной мембраны, в отдельных случаях — с небольшими участками ее утолщения или расслоения, что не позволяло исключать СА и устанавливать точный диагноз. Такая ситуация возможна в случае раннего возраста пациентов либо, возможно, мягкого течения процесса, в результате чего скорость нарастания морфологических изменений не высока, как, например, при аутосомно-доминантном варианте СА.
После первичного морфологического исследования нами был проведен сравнительный клиническо-лабораторный анализ показателей полученных групп в зависимости от патологии.
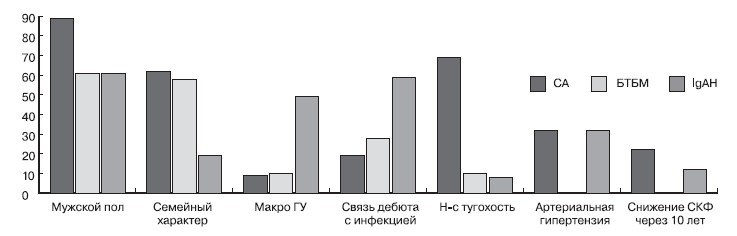
Как для СА и БТБМ, так и для IgA-нефропатии характерно преобладание лиц мужского пола: 18 (90 %), 11 (65 %) и 13 (65 %) соответственно, достоверно (р < 0,05) в сравнении с женским полом.
Для IgA-нефропатии наиболее частым первоначальным признаком болезни явилось сочетание ГУ с протеинурией до 1 г/л у 13 (65 %) пациентов с этой патологией и остается ведущим признаком в течение примерно 7 лет — до 37 %. Сопоставимо часто в тот же период встречается изолированная ГУ — до 25 %. При длительности болезни более 7 лет самым частым признаком заболевания становится сочетание ГУ с протеинурией 3 г/л и более — 50 %. Чаще, чем при других патологиях, в дебюте IgA-нефропатии встречалась макрогематурия (у 10 [50 %] обследованных) и в дальнейшем выявлялась у 9 (45 %). Развитие артериальной гипертензии отмечается у 7 (35 %) больных. У 3 (15 %) обследованных с этим заболеванием было выявлено снижение скорости клубочковой фильтрации (СКФ): через 5 лет от начала болезни частота встречаемости снижения СКФ составила 5 % (1 ребенок), через 7 лет от дебюта этот показатель соответствовал 10 % (2 детей), через 11—15 % (3 детей). На долю снижения СКФ соответствующих II, III, IV стадий хронической болезни почек (ХБП) приходилось по 1 (33%) обследованному. Для всех трех пациентов протеинурия была нефротического уровня, причем у 67 % отмечены другие признаки нефротического синдрома (гипопротеинемия, гипоальбуминемия, гиперхолестеринемия), у тех же пациентов имела место макрогематурия, и 67 % из них страдали артериальной гипертензией. Для 67 % при световой микроскопии был выявлен ФСГС, для 33 % - нефросклероз.
Картина СА характеризовалась преобладанием в дебюте изолированной ГУ и ГУ в сочетании с протеинурией до 1 г/л у 8 (40 %) и 9 (4 5%) обследованных соответственно. Среди 85 % детей с СА отмечено нарастание уровня протеинурии с течением времени. Частота ГУ в сочетании с ПР 1-3 г/л возрастает с 5 % в начале заболевания до 31 % в течение 10 лет. А частота ГУ в сочетании с ПР 3 и более г/л увеличивается за тот же промежуток времени с 5 до 50 %. Семейный характер гематурии при СА является частым признаком и обнаруживается у 13 (65 %) обследованных. Характерным признаком при СА является также нейросенсорная тугоухость, которая развилась у 14 (70 %) обследованных нами детей с этим диагнозом к 7,7 ± 5,0 годам. Развитие артериальной гипертензии наблюдается так же часто, как и при IgA-нефропатии, — у 7 (35 %) больных. Шесть (30 %) пациентов с СА течение болезни привело к снижению СКФ. Через 5 лет от начала болезни частота снижения СКФ соответствовала 10 % (2 ребенка).
Дальнейшее течение болезни сопровождается увеличением частоты снижения СКФ — до 25 % (5 детей) — через 7 лет от начала болезни и до 30 % (6 детей) через 11 лет (50 % — ХБП II стадии, 33 % — III стадии, 17 % — ХБП IV стадии из всех пациентов с СА и снижением СКФ). Для всех этих детей имела место отягощенность семейного анамнеза по заболеваниям почек, уровень протеинурии соответствовал нефротическому, хотя для 40 % из них начало заболевания характеризовалось изолированной ГУ; 80 % имели макрогематурию, нейросенсорную тугоухость. У 40 % к 15 годам развилась артериальная гипертензия. Морфологическим заключением при светооптическом исследовании в 60 % имели место склерозирующие процессы (40 % ФСГС, 20 % нефросклероз) и в 40 % — мезангио-пролиферативный гломерулонефрит.
При БТБМ частота семейного характера патологии составляет 10 (59 %) обследованных. Как и при других описанных патологиях, самым частым признаком начала болезни является сочетание ГУ с протеинурией до 1 г/л — 12 (70,5 %) пациентов. Но в отличие от СА и IgA-нефропатии этот показатель в первые 5 лет болезни возрастает до 88 %, остается неизменным и через 7 лет от начала заболевания. У 3 пациентов с признаками БТБМ по электронной микроскопии к 7 годам болезни было выявлено повышение уровня протеинурии до более 1 г/л, но менее 3 г/л и у 2 пациентов с выявленной при электронной микроскопии БТБМ к 7-му году заболевания уровень протеинурии нарос до 3 г/л и более. Не исключено, что нарастание протеинурии такого уровня объясняется тем, что из-за раннего возраста детей во время проведения им электронной микроскопии был пропущен СА, в связи с тем что к этому возрасту не успевали развиться его специфические морфологические признаки (расслоение и фрагментация базальной мембраны гломерул). Клиническая картина позволила нам пересмотреть диагноз 2 пациентов в пользу СА.
Клинико-лабораторные характеристики основных причин гломерулярной гематурии отражены на рис. 1.
Рисунок 1.Клинические признаки основных причин гломерулярной гематурии (п = 57).
После проведения клинического анализа пациентов для уточнения диагноза нами было выполнено иммуногистохимическое исследование α3- и α5-цепей коллагена в гломерулярной базальной мембране.
В результате проведенного иммуногистохимического исследования нефробиоптатов и тщательного анализа историй болезни нами было установлено, что наиболее частой причиной гломерулярной гематурии у детей является Х-сцепленный синдром Альпорта — 23 (40,5 %) обследованных, аутосомный вариант СА обнаружен у 3 (5,5 %) детей, БТБМ — у 11 (19 %) из всех обследованных (рис. 2).
Рисунок 2. Структурагломерулярной гематурии у детей в российской популяции (n=57), %
, %.")
Обсуждение исследования. Таким образом, клинико-лабораторная картина начала болезни и в течение первых пяти лет заболевания не имеет существенных различий при всех трех патологиях и не позволяет дифференцировать эти состояния, не прибегая к дополнительным методам исследования. Прогноз заболеваний различен, снижение СКФ, развитие хронической почечной недостаточности происходят и в более раннем возрасте чаще при СА, чем при IgА-нефропатии. Явления склерозирования почечной ткани сопровождают 50 % (8 детей) пациентов с явлениями снижения СКФ и 56 % (9 обследованных) с явлениями артериальной гипертензии. Артериальная гипертензия выявляется у всех пациентов со снижением СКФ. Из чего следует, что артериальная гипертензия и выявление склероза, как фокального, так и тотального, — это неблагоприятные предикторы в отношении прогноза болезни, что не противоречит данным G. D'Amico [16] и J.A. Tumlin [17].
Согласно нашим данным, в структуре причин синдрома гломерулярной гематурии у детей Российской Федерации преобладает патология коллагена IV типа (65 %), представленная Х-сцепленным синдромом Альпорта в 40,5 % случаев, аутосомным вариантом синдрома Альпорта в 5,5 % случаев и болезнью тонких базальных мембран в 19 % случаев. IgA-нефропатия является причиной гематурии в 35 % случаев. Данные по частоте патологии коллагена IV типа и IgA-нефропатии совпадают с результатами исследования C.H. Schroder et al. (1990) [18]. Дебют наиболее частых гломерулярных причин гематурии характеризуется сходной клинико-лабораторной картиной, для которой естественно преобладание ГУ в сочетании с небольшой протеинурией — не более 1 г/л — 60 % обследованных. Вторым по частоте признаком дебюта описанных болезней является изолированная ГУ — 25 %. Нередким симптомом дебюта, в большей степени характерного для IgA-нефропатии, является макрогематурия; общая частота — 25 %. Дебют заболевания с изолированной ГУ, даже в случае длительно текущей изолированной микрогематурии, не является прогностически значимым в плане благоприятности течения процесса признаком, в особенности для пациентов с СА, т. к., по нашим данным, к 10,6 ± 4,9 года жизни у 50 % из них уровень протеинурии достигает нефротического уровня, еще у 31 % уровень протеинурии соответствует 1—3 г/л и только у пятой части этих пациентов гематурия остается к этому возрасту изолированной или сочетается с протеинурией не более 1 г/л. Наличие или нарастание протеинурии до нефротического уровня является прогностически неблагоприятным признаком по отношению к развитию почечной недостаточности, которая может наступить уже к 13,0 ± 3,6 года жизни пациента (рис. 3).
Рисунок 3. Протеинурия при основных причинах гломерулярной гематурии (п - 57).
Неблагоприятным фактором является также присоединение артериальной гипертензии, которое одинаково характерно для больных СА и IgA-нефропатией (по 35 % соответственно), чаще происходит к 10 ± 3,5 годам у пациентов с СА и к 12± 3 годам у пациентов с IgA-нефропатией. Кроме того, для IgA- нефропатии характерно более позднее начало болезни, наиболее часто соответствующее 12 ± 3,5 годам, когда большая часть пациентов с СА имеет уже развитую картину процесса. Стоит учитывать, что, по нашим данным, до 15 % детей с СА, первые симптомы болезни у которых были обнаружены в 5—12 лет, не проходили регулярного медицинского обследования. Наше исследование выявило, что прогрессирование заболевания, приводящее к снижению СКФ, в т. ч. развитие хронической почечной недостаточности с необходимостью проведения заместительной терапии и почечной трансплантации, чаще встречается среди больных СА и к 15 годам соответствует 25 % обследованных. Полученные данные относительно возраста развития хронической почечной недостаточности сопоставимы с результатами исследования, проведенного М.С. Игнатовой (2006) [2], а также с многоцентровым исследованием J. Jais и соавт. (2003) [19].
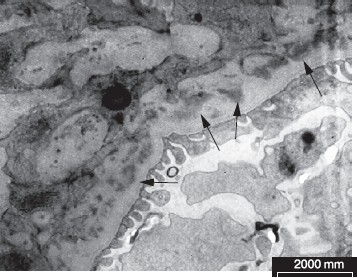
Рисунок 4. Электронная микроскопия. Парамезангиальные депозиты при IgA-нефропатии (стрелки).
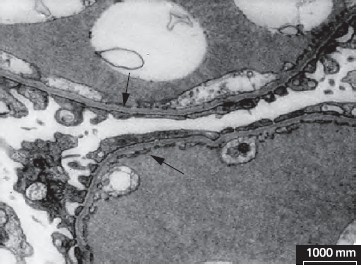
Рисунок 5. Электронная микроскопия. Равномерное истончение ГБМ при БТБМ.
Патология коллагена IV типа является ведущей причиной гломерулярной гематурии для детей в российской популяции, причем преобладающим в ней является Х-сцепленный синдром Альпорта. Клинико-лабораторная картина гематурических болезней, картина светооптической микроскопии, особенно на начальных стадиях болезни, не имеют ярких отличительных признаков, позволяющих установить диагноз, не прибегая к электронной микроскопии. С учетом высокого удельного веса болезней коллагена IV типа для улучшения качества диагностики и дифференциальной диагностики между этими болезнями морфологическое исследование должно носить полный характер с включением светооптической и электронной микроскопии, а также иммуногистохимического исследования.
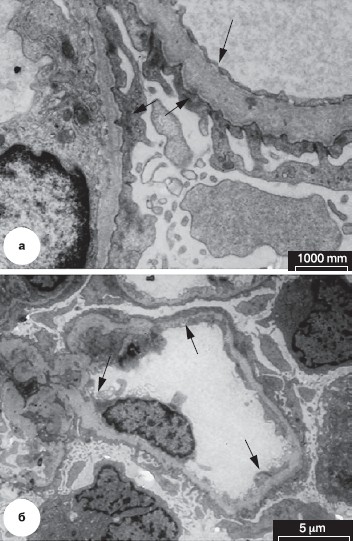
Рисунок 6. Электронная микроскопия. Синдром Альпорта.
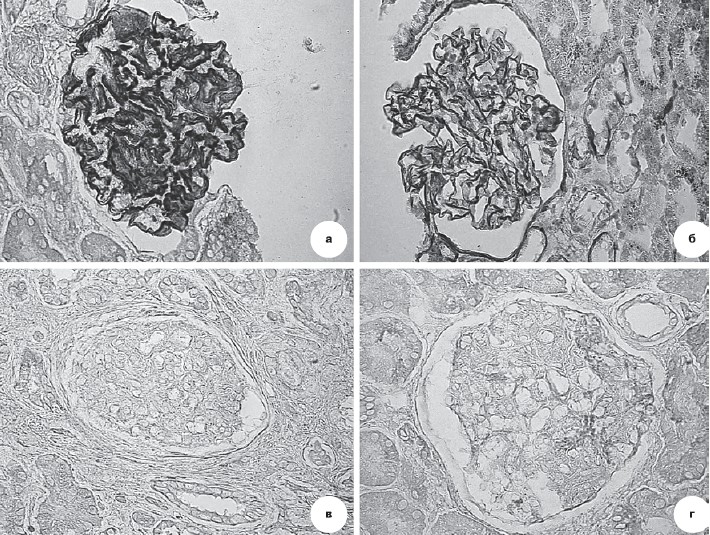
Рисунок 7. Иммуногистохимия.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}