
Атипичный гемолитико-уремический синдром (аГУС) представляет собой тяжелое системное заболевание, характеризующееся тромбоцитопенией, гемолитической анемией и острым повреждением почек, которое индуцируется гиперактивацией комплемента по альтернативному пути [1, 2]. Это очень редкое заболевание с частотой примерно 0,5 случая на млн человек в год. До появления экулизумаба (гуманизированного моноклонального антитела против С5, которое блокирует терминальный путь каскада комплемента) оно имело очень плохой прогноз [3, 4]. Являясь типичным заболеванием, опосредованным комплементом, аГУС развивается у людей с предрасполагающими генетическими аномалиями в генах комплемента после воздействия множества провоцирующих/причинных событий, включая инфекции, лекарства, злокачественные новообразования, трансплантацию и беременность [5].
Генетические исследования пациентов с клиническим диагнозом аГУС выявляют мутации в генах, связанных с альтернативным путем активации системы комплемента, почти в половине всех случаев [6, 7]. Список широко известных генов при аГУС включает CFH (причастен примерно у 25% пациентов), CD46 или MCP (около 10%), C3 (около 6%), CFI (около 6%), CFB (около 2%), THBD (около 2%) и не связанный с комплементом ген диацилглицеролкиназы эпсилон – DGKE (приблизительно 3%) [5]. Аутоантитела к фактору H обнаруживаются в 5–13% случаев и связаны с отсутствием обеих копий CFHR1 [8]. Если обнаруживается генетическая вариация, ее часто считают предрасполагающим фактором, а не непосредственной причиной аГУС. Это различие отражает высокую вариабельность пенетрантности болезни, за исключением патогенных вариантов DGKE, которые подчиняются аутосомно-рецессивному типу наследования [9].
При выявлении мутации генов у пациента с аГУС крайне важно определить их клиническую значимость, т.к. это имеет отношение к длительности антикомплементарной терапии. Выявляемые варианты мутаций в генах, связанных с альтернативным путем активации комплемента, не во всех случаях имеют отношение к патогенезу аГУС. В ряде публикаций показано, что ультраредкие, но доброкачественные варианты не патогенны [10]. Например, M.C. Marinozzi et al. экспериментально продемонстрировали, что 9 из 15 зарегистрированных мутаций гена CFB не были связаны с патогенезом аГУС [11]. Также были идентифицированы новые варианты мутаций CFH, которые могут не быть связанными с аГУС [12]. Эти отчеты подтверждают ценность функциональных исследований для оценки влияния разных генетических вариантов, достаточно трудоемких и сложных, что делает функциональную оценку непрактичной в каждом случае.
Цель исследования: изучение спектра генетических мутаций у пациентов с аГУС.
Материал и методы
На базе Научно-практического центра нефрологии и патологии трансплантированной почки ГКБ № 52 проведено ретроспективное исследование, в которое были включены 44 пациента (28 мужчин и 16 женщин) с аГУС, наблюдавшихся в клинике с 2014 по 2021 г. Средний возраст пациентов составил 29,6±11,1 года.
Все пациенты с установленным диагнозом аГУС условно были разделены на 4 группы. 1-ю представляли 7 пациенток с акушерским аГУС. Во 2-ю группу входили 11 пациентов с аГУС, диагностированным в собственных почках, не нуждавшихся в заместительной почечной терапии (ЗПТ) на момент последнего наблюдения. Третья группа представлена 10 пациентами с аГУС и терминальной хронической почечной недостаточностью (ТХПН) на лечении программным диализом. Некоторым пациентам этой группы диагноз аГУС окончательно верифицирован после проведения генетического исследования, т.к. клинически течение заболевания почек, приведшего к ТХПН, заставляло подозревать аГУС. В связи с планируемой трансплантацией почки у пациентов данной группы генетический анализ стал ключевым для определения тактики их ведения в посттрансплантационном периоде. В 4-ю группу вошли 16 пациентов с аГУС после аллотрансплантации трупной почки (АТП).
Пациентам всех групп проведены физикальный осмотр, лабораторно-инструментальное обследование (клинический и биохимический анализы крови, общий анализ мочи и суточный анализ мочи, электрокардиография, хокардиография, ультразвуковое исследование почек и органов брюшной полости). Ряду пациентов по показаниям дополнительно проведены мультиспиральная компьютерная томография и магнитно-резонансная томография головного мозга, эзофагогастродуоденоскопия и фиброгастродуоденоскопия. Пункционная биопсия почки с проведением световой микроскопии и иммунофлуоресцентного исследования на замороженных срезах была выполнена 25 (57%) пациентам. У всех 25 пациентов по результатам биопсии почки выявлены признаки тромботической микроангиопатии (ТМА). По клинической картине и результатам лабораторно-инструментальных методов исследования пациентам проводилась оценка наличия поражения органов-мишеней помимо почек в рамках аГУС (сердце, головной мозг, кишечник и другие органы).
Всем пациентам выполнен генетический анализ крови на панель аГУС. ДНК выделялась с помощью стандартных методик наборами реагентов Qiagen на системах автоматического выделения ДНК QIAcube и Freedom EVO. Качественные и количественные показатели образца ДНК измерялись с помощью гель-электрофореза нуклеиновых кислот и с интеркалирующим красителем на флуориметре Qubit. ДНК ферментативно разрезали на фрагменты определенной длины, к которым добавляли специальные последовательности, называемые адаптерами. Библиотеки ДНК готовили с использованием роботизированной станции Tecan Freedom EVO и амплификатора T100. Далее оценивали количество и качество созданной библиотеки ДНК. Качественный анализ выполняли методом капиллярного гель-электрофореза на приборах Bioanalyzer 2100 (Agilent) или LabChip GX Touch (Perkin Elmer). Количественный анализ проводили методом полимеразной цепной реакции (ПЦР) в реальном времени на амплификаторе StepOnePlus. Необходимые для последующего анализа последовательности генома обогащали с помощью биотинилированных РНК-зондов и анализировали на приборах HiSeq 2500 и MiSeq по протоколу производителя. После контроля качества полученных данных (точность прочтения, глубина и полнота прочтения) проводили поиск генетических вариантов. В качестве референсного генома использовали сборку GRCh38.p13. Каждому обнаруженному варианту присваивали одну из пяти категорий патогенности или доброкачественности согласно рекомендациями ACMG. Врач-генетик составлял список генетических вариантов, связанных с клиническими симптомами пациента на основании данных клинической картины или направительного диагноза пациента.
Выделение ДНК, приготовление и секвенирование ДНК-библиотек
Выделение ДНК проводили с помощью набора «QIAmp DNA mini kit» (Qiagen). Молекулярную массу геномной ДНК подтверждали, используя гель-электрофорез, при этом следили за отсутствием деградации ДНК и загрязнения РНК. Концентрацию выделенной ДНК определяли, используя Qubit 3.0 (Life Technologies). ДНК-библиотеки готовили с использованием набора NEBNext® Ultra™ DNA Library Prep Kit for Illumina (New England Biolabs), используя адаптеры последовательности для секвенирования на приборах Illumina, согласно стандартному протоколу, предложенному производителем. Двукратное индексирование выполняли с помощью ПЦР с NEBNext Multiplex Oligos for Illumina® (Dual Index Primers Set 1). Качество и концентрацию библиотек определяли с помощью Bioanalyzer 2100 (Agilent Technologies). Для обогащения фракции геномной ДНК использовали набор The SureSelect XT2 Target Enrichment System (Agilent Technologies). ДНК-библиотеки секвенировали на геномном анализаторе HiSeq2500 (Illumina) с использованием парных чтений с длиной чтения 100 нуклеотидов.
Биоинформатический анализ
Нуклеотиды с 3’-конца, имеющие качество прочтения ниже 10, обрезались с помощью программы cutadapt. Выравнивание на референсный геном hg19 (GRCh37.p13) было выполнено с помощью bwa. Дедупликация ридов выполнена с помощью samtools rmdup. Контроль качества чтений осуществляли с помощью программ FastQC и Genotek quality analysis solution. Распознавание коротких вариантов (изменений, мутаций) осуществляли GATK HaplotypeCaller, согласно установленному протоколу GATK DNA-seq. Для рассмотрения выбирали лишь те варианты, покрытие в которых составляло ≥12. Влияние каждого изменения оценивали с помощью snpEff, оценки патогенности и консервативности, данные для которых извлекали из баз данных dbNSFP, Clinvar, OMIM и HGMD, а также с использованием утилит SIFT и polyphen2 для предсказания возможной патогенности мутации. Информацию о частоте мутаций брали из 1000 Genomes project, ExAC и Genotek frequency data. Аннотацию мутаций и их патогенность предсказывали, согласно стандартам и руководству, разработанным ACMG, AMP и CAP для интерпретации мутаций, полученных с помощью секвенирования. Число копий мутаций и цитогенетические перестановки осуществляли с помощью CNVkit.
Обнаруженные у пациентов мутации были условно разделены на 3 группы: мутации, характерные для аГУС (CFH, CFI, C3, CD46 (MCP), THBD, DGKE, CHFR1/CHFR3), мутации тромбофилий (MTHFR, ITGA2, ITGB3, F5, F7, F13A1, FGB) и мутации ADAMTS13.
Статистические расчеты (описательная статистика) выполнены с помощью программы Excel и статистической программы SPSS Statistics 23.
Результаты
Среди пациентов с аГУС мутации в системе комплемента обнаружены у 30 (68%) из 44 пациентов. Мутации тромбофилий выявлены у 14 (32%) пациентов. При этом у 11 (25% от общего числа) пациентов наблюдалось сочетание мутаций в системе комплемента с мутациями тромбофилий. Изолированные мутации в системе комплемента обнаружены у 19 (43%) пациентов, изолированные мутации тромбофилии – у 2 (5%) . У 6 (14%) пациентов выявлены мутации ADAMTS13 (у 2 пациентов изолированно, у 3 – в сочетании с мутациями системы комплемента и тромбофилий, у 1 – в сочетании с мутациями тромбофилий). У 9 (20%) пациентов мутации не обнаружены (рис. 1).
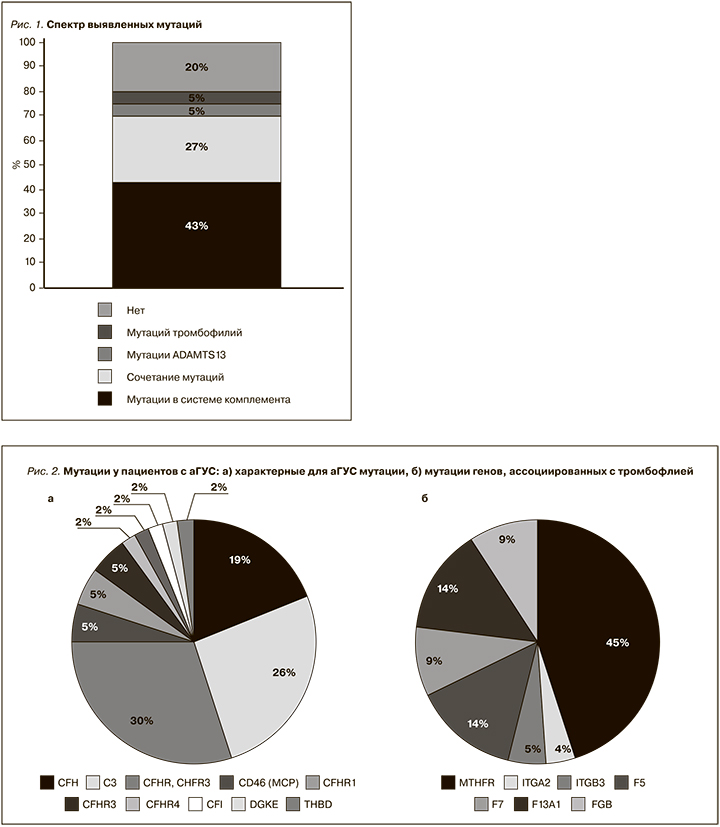
Среди характерных для аГУС мутаций чаще всего встречались мутации генов CFHR1, CFHR3 – у 13 (30%) пациентов. При этом чаще наблюдалось именно сочетание CFHR1, CFHR3. Изолированные мутации в этой группе наблюдались значительно реже: CFHR1 – 2 (5%) пациента, CFHR3 – 2 (5%), CFHR4 – 1 (2%) и CFHR5 – 1 (2%) пациент. Вторыми по частоте у наших пациентов были мутации в генах С3 – 11 (26%) пациентов. У 8 (19%) пациентов были обнаружены мутации в генах CFH. У 2 (5%) пациентов выявлены мутации CD46 (MCP). Остальные мутации встречались значительно реже (рис. 2а).
Помимо известных патогенных для аГУС мутаций проведен анализ других мутаций (чаще всего ассоциированных с тромбофилией), обнаруженных у наших пациентов с аГУС (рис. 2б). В большинстве случаев это были мутации MTHFR – 10 (45%) пациентов. У 14% пациентов были мутации F5 и F13А1, реже встречались мутации FGB и F7 (по 9%), ITGB3 (5%) и ITGA2 (4%).
Также был проведен анализ различных мутаций и поражений органов-мишеней по выделенным группам пациентов (рис. 3). У 5 из 7 пациенток с акушерским аГУС обнаружены характерные для аГУС генетические мутации (CFH, CFI, C3, CD46, THBD), при этом у трех из них эти мутации сочетались с тромбофилическими мутациями и мутациями ADAMTS13 (рис. 3а). Чаще всего у пациенток с акушерским аГУС наблюдалось изолированное поражение почек, но было по 1 пациентке с поражением сердца, кишечника или центральной нервной системы (ЦНС). Биопсия почки выполнялась только одной пациентке и подтвердила наличие ТМА. Шесть из семи пациенток данной группы начали своевременное лечение экулизумабом. При этом у 4 пациенток функция почек полностью восстановилась, 2 из них имели мутации в генах CHFR1, CHFR3, CD46 и С3, у 2 других патогенных мутаций не было. У 1 пациентки почечная функция восстановилась до уровня начальной ХПН, у нее были мутации CHFR1, CHFR3 и THBD. Одна пациентка с мутациями CHFR1, CHFR3 осталась на лечении программным гемодиализом. Единственной пациентке данной группы, которой не было своевременно начато лечение экулизумабом, диагноз аГУС установлен еще в период, когда лечение экулизумабом в нашей стране было не доступно. Ей была выполнена АТП, однако через 2 года в связи с рецидивом аГУС в трансплантате она вернулась на лечение программным гемодиализом. У нее были обнаружены мутации генов CFH и CFI. При повторной трансплантации назначена терапия экулизумабом, на фоне чего почечный трансплантат удовлетворительно функционирует в течение 6,5 года.
Среди 11 пациентов с аГУС в собственных почках без ЗПТ у 6 были обнаружены патогенные мутации, которые в большинстве случаев (у 4 пациентов) представлены мутациями CFHR1, CFHR3 (рис. 3б). В данной группе нередко наблюдалось сочетание поражения почек с поражением кишечника и ЦНС (по 4 пациента), сердца (3 пациента). Четырем пациентам выполняли биопсию почки, подтвердившую ТМА. Все пациенты данной группы получают лечение экулизумабом. На фоне лечения экулизумабом 3 из 11 пациентов полностью восстановили функцию почек. Среди этих 3 пациентов у двух были мутации CFHR1, CFHR3, у одного патогенных мутаций обнаружено не было. Средний уровень креатинина крови остальных 8 пациентов составил 211±140 мкмоль/л.
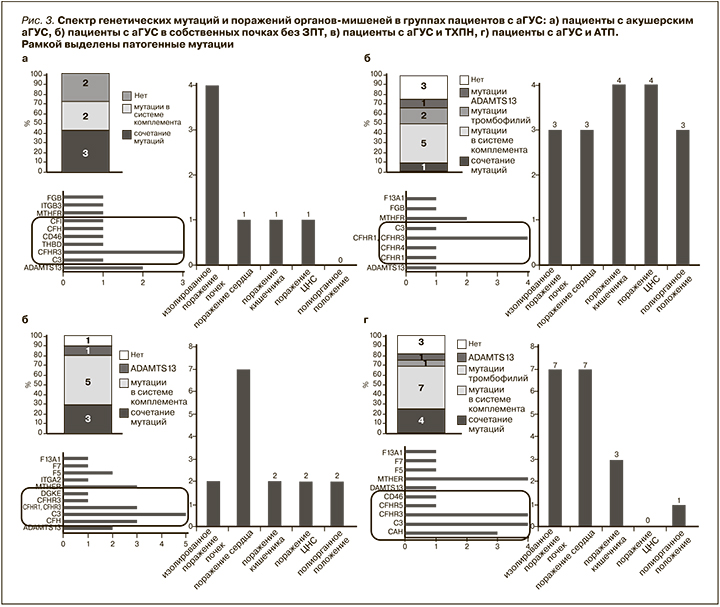
В группе пациентов с аГУС и ТХПН у 8 из 10 пациентов обнаружены патогенные мутации (рис. 3в). В большинстве случаев это были мутации С3, CFH и CFHR1, CFHR3. Нередко наблюдалось сочетание патогенных мутаций с мутациями ADAMTS13 и тромбофилий, среди которых чаще всего встречались мутации MTHFR и F5. У 7 пациентов представленной группы наблюдалось поражение сердца в рамках аГУС, реже выявлялись поражения кишечника и ЦНС. Восьми из 10 пациентов выполнена биопсия почки, подтвердившая ТМА. Пять из 10 пациентов получают терапию экулизумабом, обусловленную наличием у них тяжелого поражения других органов-мишеней в рамках аГУС (сердце, ЦНС). Остальным пациентам планируется лечение экулизумабом после выполнения АТП.
Патогенные мутации в группе аГУС и АТП выявлены у 11 из 16 пациентов (рис. 3г). При этом чаще всего обнаруживали мутации С3, CFHR1, CFHR3 и CFH, как и в группе пациентов с аГУС и ТХПН. Нередко наблюдалось сочетание с мутациями MTHFR. В данной группе среди поражения органов-мишеней преобладало поражение сердца (выявлено у 7 пациентов), как и в группе с аГУС и ТХПН. Диагноз ТМА подтвержден по биопсии собственных почек у 3 пациентов, у 4 – по биопсии трансплантированной почки. Все 16 пациентов получают лечение экулизумабом. Среди этих 16 пациентов 10 диагноз поставлен еще до выполнения АТП и терапию экулизумабом они начали получать сразу после операции. У этих пациентов выявлялись мутации CFH, CFI, C3, CD46 (MCP), CFHR1, CFHR3, CFHR5.
К настоящему времени у всех этих пациентов трансплантаты функционируют и средний уровень креатинина крови в данной группе пациентов составляет 125±29 мкмоль/л. Остальным 6 пациентам диагноз аГУС поставлен уже после АТП. Трое из пациентов, у которых определялись мутации CFHR1, CFHR3, CFH, C3, вернулись на лечение программным диализом, несмотря на проводимую антикомплементарную терапию.
У оставшихся 3 пациентов с мутациями CFHR1, CFHR3, С3 средний уровень креатинина крови составляет 208±105 мкмоль/л.
Обсуждение
Выявление патогенного генетического варианта у пациента с аГУС подтверждает диагноз и позволяет с точностью установить причину заболевания, облегчая ведение пациента, эффективное лечение и генетическое консультирование.
Скрининг редких вариантов генов комплемента обычно включает секвенирование не менее пяти генов комплемента – CFH, C3, CFI, CFB и MCP, а также выявление гибридных генов CFHR1-CFHR3, вызывающих неаллельные гомологичные рекомбинации [13]. На сегодняшний день у пациентов с аГУС идентифицировано более 500 вариантов этих пяти генов комплемента [14]. Эти гены кодируют белки, которые участвуют в регуляции как на клеточной поверхности, так и в жидкой фазе альтернативного пути комплемента, за исключением MCP, который участвует только в регуляции на клеточной поверхности альтернативного пути комплемента [15]. Результаты исследований, в ходе которых проведен скрининг редких вариантов гена комплемента более чем у 3000 пациентов во всем мире, показали, что аГУС является заболеванием, опосредованным комплементом. Примечательно, что во всех опубликованных сериях примерно половина обнаруженных вариантов была локализована в гене CFH [14].
Так, в итальянском исследовании M. Noris et al., включившем 273 пациента с аГУС (73% пациентов из Европы, 16% из Америки, 2% из Африки, 1% из Азии, 8% с Ближнего Востока) в период с 1996 по 2007 г. мутации в системе комплемента обнаружены примерно у 70% пациентов. При этом чаще всего наблюдались мутации CFH – 23%, реже мутации MCP – 7%, THBD – 5%, C3 – 4% и CFI – 4%. Пациенты с мутациями CFH или THBD имели самое раннее начало и самую высокую смертность. Мутации MCP связаны с лучшим прогнозом. Плазмотерапия индуцировала ремиссию в 55–80% случаев у пациентов с мутациями CFH, C3, THBD или аутоантителами, в то время как пациенты с мутациями CFI плохо реагировали на лечение [9].
Во французском исследовании среди 214 пациентов с аГУС, наблюдавшихся с 2000 по 2008 г., в 60,2% случаев выявлены мутации в системе комплемента. Среди них чаще всего также встречались мутации CFH – 28%, реже мутации MCP – 9%, CFI – 8%, С3 – 8% и CFB – 2% [16].
В американском исследовании 144 пациентов с аГУС имелось схожее распределение по мутациям: CFH – 27%, CFI – 8%, MCP – 5%, CFB – 4%, THBD – 3% и C3 – 2% [17].
Несколько иное распределение частоты мутаций получилось в японском исследовании M. Fujisawa et al. В исследование были включены 118 японских пациентов с 1998 по 2016 г. У них наиболее частыми (31%) генетическими аномалиями были мутации C3, а частота мутаций CFH относительно низкой (10%) по сравнению с западными странами. В этой когорте преобладал вариант С3 p.I1157T (23%), что было связано с благоприятными исходами, несмотря на частые рецидивы. В общей сложности 72% пациентов получали плазмотерапию, 42% лечили экулизумабом. Прогноз японских пациентов с аГУС был относительно благоприятным с общей смертностью 5,4% и почечной смертностью 15% [18].
В нашем исследовании мутации в системе комплемента обнаружены у 68% пациентов с аГУС, что соотносится с международными данными. По спектру выявленных мутаций у наших пациентов чаще (26%) наблюдались мутации С3, что больше соответствует данным пациентов японской группы. С другой стороны, частота обнаружения мутаций CFH (19%) скорее ближе к результатам западных исследователей, хоть и представлена несколько более низким процентом.
В группе пациенток с акушерским аГУС мутации в системе комплемента наблюдались у 71% пациенток. В работе F. Fakhouri et al. проведен анализ данных 21 пациентки с акушерским аГУС: мутации в системе комплемента обнаружены у 86%, при этом чаще, чем в общей популяции, выявлялись мутации CFH (45%) [19]. В нашей группе пациенток с акушерским аГУС мутации CFH, CFI, C3, CD46, THBD наблюдались с одинаковой частотой – 14%, что, вероятно, обусловлено небольшим числом пациенток.
У пациентов с аГУС с поражением собственных почек без ЗПТ не было обнаружено серьезных мутаций CFH, CFI, которые, как известно, значимо ухудшают прогноз и часто приводят к ТХПН. Однако у этих пациентов нередко наблюдались экстраренальные проявления с поражением кишечника, ЦНС и сердца.
Интересно, что пациенты групп ТХПН и АТП по своему спектру генетических мутаций и экрстраренальным проявлениям очень схожи. Это обусловлено тем, что по своей сути пациенты из группы ТХПН постепенно переходят в группу пациентов с АТП, и наоборот все пациенты с АТП еще недавно были пациентами с ТХПН.
При аГУС сообщается об изменениях генов CFHR1, CFHR3 и фактора H в сочетании с интактными генами CFHR2, CFHR4 и CFHR5. А изменения в каждом из пяти генов CFHR в контексте интактного гена фактора H описаны при С3-гломерулопатии [20]. Белки CFHR с 1 по 5 структурно сходны с фактором H и кодируют области, расположенные рядом с геном CFH на хромосоме 1. Гены CFHR, считающиеся псевдогенами, имеют высокую степень гомологии последовательностей с CFH, следовательно, восприимчивы к генетическим перестройкам, приводящим к образованию гибридов CFH-CFHR. При этом их пенетрантность составляет около 50% как в семейных, так и в спорадических случаях, и для развития заболевания у людей с данными мутациями требуется серьезный триггер [21].
Гомозиготная делеция CFHR3-CFHR1 наблюдается примерно у 15% преимущественно молодых пациентов с аГУС. У этих пациентов часто обнаруживаются аутоантитела к фактору Н. Точные механизмы, посредством которых при аГУС гомозиготный дефицит CFHR1 и CFHR3 индуцирует выработку антител, неясны. Один из механизмов заключается в том, что FHR3 связывается с фрагментом C3d и блокирует адъювантный эффект C3d при активации В-клеток [22].
В индийской статье S. Kandari et al. [23] проанализированы результаты генетического исследования 17 пациенток с акушерским аГУС. У 11 пациенток обнаружены гетерозиготные делеции CFHR1 и CFHR3, у 4 – гомозиготные делеции CFHR1 и CFHR3. И лишь у 2 пациенток мутаций не было.
Согласно нашим данным, у пациенток с акушерским аГУС мутации CFHR1, CFHR3 также являлись наиболее частой генетической аномалией в данной группе – у 3 из 7 пациенток. Как известно, беременность и роды являются очень серьезным триггером, что, по всей видимости, обусловливает высокую частоту встречаемости данной аномалии у пациенток с акушерским аГУС. Кроме того, мутации CFHR1 и CFHR3 нередко наблюдались и в остальных группах пациентов с аГУС.
К сожалению, по техническим причинам не всем этим пациентам выполнен анализ крови на антитела к фактору Н. Среди 4 обследованных пациентов с подобными генетическими аномалиями у 2 б обнаружены антитела к фактору Н. Вклад тромбофилических мутаций в развитии аГУС не установлен. Однако, как следует из представленных данных пациентов с аГУС, помимо мутаций в системе комплемента нередко можно обнаружить тромбофилические мутации. В литературе описаны мутации при аГУС, не связанные с системой комплемента. Примером служат мутации в генах диацилглицеролкиназы эпсилон (DGKE), инвертированного формина-2 (INF2) и плазминогена (PLG). У наших пациентов встречались мутации DGKE, а вот мутации INF2 и PLG обнаружены не были, что обусловлено все-таки достаточно редкой встречаемостью этих аномалий.
Внепочечные проявления аГУС наблюдаются примерно у 20% пациентов [24]. При этом могут поражаться многие системы органов, в т.ч. периферическая и центральная нервная системы (8–48%), желудочно-кишечный тракт (8–10%), сердечно-сосудистая система (3–10%), кожа, легкие (около 21%), глаза (4%). Некоторые из этих проявлений возникают в острой фазе аГУС, другие могут быть проявлением отдаленных последствий бесконтрольной активации комплемента.
Согласно нашим данным, у пациенток с акушерским аГУС в большинстве случаев наблюдалось изолированное поражение почек. У пациенток с аГУС в собственных почках без ЗПТ среди экстраренальных проявлений чаще встречались поражения кишечника и ЦНС (по 36%). А в группах ТХПН и АТП экстраренальные проявления были схожими и чаще представлены поражением сердца (70 и 44% соответственно). Поражение сердца у этих пациентое может как быть прямым следствием активации комплемента и ТМА, так и усугубляться на фоне тяжелой артериальной гипертензии и уремии [5], которые наблюдаются у пациенток с ТХПН и через которые проходят все пациентки с АТП.
В группе пациентов после АТП вне зависимости от выявленных мутаций исходы по большей части были связаны со своевременностью начала терапии экулизумабом. Так, у пациентов, которым лечение экулизумабом начато сразу после АТП в качестве профилактической терапии, почечные трансплантаты сохранили свою функцию, несмотря на наличие таких мутаций, как CFH, CFI, C3, которые, как известно, определяют частые рецидивы аГУС в трансплантате в отсутствие антикомплементарной терапии [25, 26]. Среди пациентов, которым лечение экулизумабом начато при развитии рецидива аГУС в почечном трансплантате, половина из них потеряли свои трансплантаты и вернулись к лечению диализом. При этом генетические мутации у этих пациентов были схожими (в т.ч. мутации CFH, C3), что подтверждает необходимость своевременной диагностики аГУС (по возможности – до выполнения АТП) и раннего начала комплемент-блокирующей терапии пациентов этой группы.
Заключение
Исследование сотен пациентов с аГУС предоставило информацию о генетических причинах болезни и корреляциях генотипа и фенотипа, которые могут прогнозировать прогрессирование заболевания, ответ на терапию и риск рецидива после трансплантации. Это позволяет осуществлять индивидуальный подход к ведению пациентов и лечению, основанному на экспертной интерпретации генетических профилей, что требует проведения генетического скрининга каждому пациенту. Задержки в получении результатов генетических или молекулярно-диагностических исследований не должны препятствовать постановке клинического диагноза и началу лечения, т.к. ранняя антикомплементарная терапия имеет решающее значение для сохранения функции почек и предотвращения необратимых последствий.


