
Диабетическая нефропатия (ДН) – одно из самых частых, тяжелых и прогностически неблагоприятных осложнений сахарного диабета (СД). В последние годы ДН заняла лидирующие позиции среди причин терминальной почечной недостаточности в индустриально развитых странах. Неудовлетворительные результаты лечения ДН связаны со сложностью патогенеза, длительным бессимптомным течением, поздней диагностикой. Проведенные в разных регионах России выборочные эпидемиологические исследования свидетельствуют, что среди больных СД фактическая распространенность поражения почек превышает регистрируемую в 2–3 раза [1].
В настоящее время не вызывает сомнения, что структурные и функциональные изменения почек появляются в первые годы течения СД. Наиболее ранние признаки почечного поражения включают гиперфильтрацию и клубочковую гипертрофию, далее развиваются утолщение и нарушение структуры базальных мембран, экспансия мезангия клубочков, перигломерулярный склероз, дистрофические и атрофические изменения канальцев, фиброз интерстиция, артерий и артериол [2, 3].
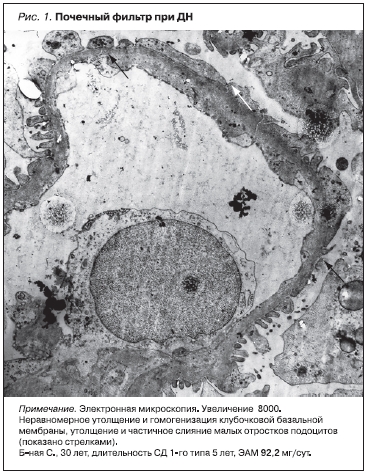
По нашим данным, выраженные изменения гломерулярного барьера, начальные признаки фиброзирования клубочков, интерстиция и сосудов почек у больных СД типа 1 (СД1) обнаруживаются уже на стадии микроальбуминурии (рис. 1, 2). На доклинических этапах ДН при нормальной экскреции альбумина с мочой (ЭАМ) и микроальбуминурии в почечных клубочках начинают накапливаться минорные (IV и VI типы) и интерстициальные (I и III типы) коллагены. В базальных мембранах канальцев и в интерстиции почек возрастает количество коллагена III, IV, в меньшей степени – I и VI типов [4, 5]. Это соответствует экспериментальным данным об активирующем влиянии гипергликемии на синтез коллагенов в клубочковых и канальцевых клетках [6], свидетельствует о раннем начале формирования фиброза почек при СД.
Появление протеинурии у большинства больных СД подтверждает выраженное и необратимое поражение почек [7]. Напротив, более ранние стадии диабетической нефропатии могут подвергаться обратному развитию. В ставшей классической работе P. Fioretto и соавт. показано, что начальные структурные изменения клубочков и интерстиция почек у больных СД1 с микроальбуминурией постепенно (в течение 10 лет) регрессируют при полном устранении гипергликемии после трансплантации поджелудочной железы [8, 9]. Использование современных технологий лечения позволило значительно снизить частоту развития поздних стадий ДН в ряде специализированных центров США, Швеции, Дании [10], а также России [8]. Эти данные указывают на то, что ранняя диагностика и лечение поражения почек имеют принципиальное значение для улучшения прогноза пациентов с ДН. В связи с этим продолжается поиск диагностических маркеров, которые бы отражали наиболее ранние стадии данного осложнения.
Альбуминурия. Выявление микроальбуминурии является стандартным методом диагностики ДН в клинической практике. Нижний порог диапазона микроальбуминурии (30 мг/сут, или 20 мкг/мин) заведомо выше, чем максимальные значения ЭАМ у здоровых лиц. Показано, что у здоровых взрослых альбуминурия обычно не превышает 15 мкг/мин, составляя в среднем около 5 мкг/мин [11]. В свою очередь наличие “высокой нормальной” ЭАМ у больных диабетом является предиктором развития микроальбуминурии [12–14]. Микро- и макроальбуминурия не представляют собой дискретные в клиническом отношении понятия, скорее их можно рассматривать как части континуума: повторные измерения ЭАМ у одних и тех же пациентов могут давать результаты в смежных диапазонах [15].
При оценке результатов определения альбуминурии у больных СД следует учитывать, что к увеличению ЭАМ могут приводить кетоацидоз, высокобелковая диета, чрезмерная физическая нагрузка, хроническая сердечная недостаточность, лихорадка, а также любое сопутствующее заболевание почек. В связи с этим определение микроальбуминурии нужно проводить после устранения кетоацидоза, инфекции мочевыводящих путей, перегрузки белком, на фоне нормальной температуры тела и обычной физической активности пациента. Для определения микроальбуминурии целесообразно использовать суточную порцию мочи. При использовании разовых (утренних) порций мочи необходимо пересчитывать альбуминурию на величину экскретируемого креатинина [15].
Средняя частота развития микроальбуминурии среди больных СД1 составляет 2,5–5,0 % в год. Большинство случаев микроальбуминурии выявляется при длительности заболевания от 5 до 20 лет, при дальнейшем увеличении длительности СД число новых случаев снижается [15]. Кумулятивная частота развития микроальбуминурии при длительности СД1 40 лет составляет 25 % [16]. При СД2 микроальбуминурия может регистрироваться уже в момент диагностики СД, что связано с длительным бессимптомным течением данной формы заболевания, частым наличием сопутствующей артериальной гипертензии и других нефропатий [15].
Распространенность микроальбуминурии при СД варьируется в различных популяциях и зависит от возраста пациентов, длительности диабета, качества контроля гликемии, наличия артериальной гипертензии и адекватности ее коррекции, этнических факторов и различий в протоколах исследования мочи [15]. Риск развития микроальбуминурии повышается с ростом уровня гликированного гемоглобина (HbA1c) и артериального давления [12, 16]. Дополнительное значение имеют дислипидемия [17, 18], наличие ретинопатии, курение [19], генетические факторы [20]. Дебют СД до пубертатного периода уменьшает риск повышения альбуминурии [17].
Российские национальные стандарты оказания помощи больным СД определяют необходимость проведения ежегодного скрининга на микроальбуминурию всем больным с момента постановки диагноза [21]. Исключение, согласно данным стандартам, представляют пациенты с СД1, заболевшие в раннем детском и постпубертатном возрасте. У этой категории больных предлагается начинать скрининг спустя 5 лет после постановки диагноза. Эта рекомендация вряд ли является оправданной в отношении больных, заболевших после пубертата. Наши данные [5] и исследования других авторов [16] свидетельствуют о нередком появлении микроальбуминурии при длительности СД1 до 5 лет при начале заболевания после пубертатного возраста.
В целом больные СД с микроальбуминурией имеют более выраженные структурные изменения в почках по сравнению с пациентами с нормальной ЭАМ. В частности, это касается толщины базальных мембран и объема мезангия, хотя четкую границу по этим параметрам между пациентами с нормо- и микроальбуминурией провести невозможно [22]. По нашим данным, фиброз отдельных клубочков, атрофия канальцев, интерстициальный фиброз обнаруживаются преимущественно у больных СД1 с микроальбуминурией, но не у пациентов с нормальной ЭАМ [23]. При СД2 выраженность структурных изменений в почках в меньшей степени соответствует альбуминурии [22]. У трети больных СД2 с микроальбуминурией выявляется нормальная структура почек (особенно при световой микроскопии), еще у трети преобладают изменения сосудов и тубулоинтерстиция [2].
Представления об эволюции микроальбуминурической стадии ДН существенно изменились в последние годы. Результаты ранних работ свидетельствовали о том, что примерно у 80 % больных СД1 c микроальбуминурией в дальнейшем развивается выраженная ДН [24, 25]. Согласно более поздним исследованиям, только у 15–45 % больных с микроальбуминурией развивается постоянная протеинурия, у 25–58 % больных ЭАМ переходит в нормальный диапазон, у остальных микроальбуминурия сохраняется [26–28]. На риск трансформации микроальбуминурии в протеинурию у больных СД1 влияют уровень HbA1c и величина исходной ЭАМ [26]. Возврату ЭАМ в нормальный диапазон способствует достижение оптимального контроля гликемии, а также применение ингибиторов АПФ [29, 30].
Результаты исследований, проведенных в последние 20 лет, убедительно показали, что микроальбуминурия является не только фактором риска прогрессирования ДН, но и предиктором сердечно-сосудистой заболеваемости. В исследовании HOPE (Heart Outcomes Prevention Evaluation study) показано, что микроальбуминурия сопряжена с повышением вероятности сердечно-сосудистых осложнений и смерти в среднем в 2, а риск развития застойной сердечной недостаточности более чем в 3 раза у больных СД2 с факторами сердечно-сосудистого риска [31]. По данным других проспективных исследований, наличие микроальбуминурии удваивает риск сердечно-сосудистых катастроф и смерти от сердечно-сосудистых причин у больных СД2 [32, 33]. При СД1 микроальбуминурия также ассоциирована с сердечно-сосудистой смертностью [34]. По данным проспективного исследования FinnDiane (Finnish Diabetic Nephropathy study), наличие микроальбуминурии ассоциировано с увеличением общей смертности больных СД1 в 2,8 раза [35].
Неблагоприятное прогностическое значение может иметь даже “высокая нормальная” ЭАМ. Установлено, что повышенный риск нефропатии и сердечно-сосудистых катастроф имеют больные с СД2, у которых ЭАМ находится в пределах верхней трети нормального диапазона (20–30 мг/сут) [36]. Для прогноза развития ДН имеет значение не только исходная величина ЭАМ, но и темпы ее повышения внутри нормального диапазона в первые годы болезни [37].
Значение микроальбуминурии как универсального маркера неблагоприятного прогноза объясняется ее тесной связью с другими факторами риска сердечно-сосудистых заболеваний [38].
Маркеры обмена внеклеточного матрикса. Центральную роль в склерозировании почек при СД играют нарушения обмена компонентов внеклеточного матрикса, прежде всего коллагенов и протеогликанов. С патологическим накоплением матрикса связано развитие гломерулярного и интерстициального фиброза. Поэтому показатели обмена матрикса изучаются в качестве потенциальных маркеров ДН.
На роль маркера диабетической нефропатии может претендовать динамика мочевой экскреции коллагена и его метаболитов. О состоянии обмена коллагена можно судить по содержанию различных типов коллагена и их метаболитов в моче. В ряде работ показано увеличение экскреции коллагена IV типа у больных СД с нефропатией [39–41]. Установлено, что мочевая экскреция коллагена взаимосвязана со степенью морфологических изменений клубочков, канальцев и интерстиция почек, отражает накопление коллагена в этих структурах [40]. По некоторым данным, содержание коллагена IV типа в моче в большей степени, чем альбуминурия, отражает процесс аккумуляции коллагена в почках [40] и более тесно связано с их фильтрационной функцией [39].
Другой подход к определению коллагена в моче у больных СД основан на выявлении продуктов обмена коллагена. Общепризнанным маркером коллагеновых белков и продуктов их расщепления является гидроксипролин. Увеличение экскреции пептидносвязанного и свободного гидроксипролина обнаружено у больных СД1 с микро- и макроальбуминурией [4].
Активность коллагенолитических ферментов и их ингибиторов в крови и моче также может быть использована для диагностики ДН. Важная роль изменений активности коллагенолитических ферментов из группы матриксных металлопротеаз (ММРs) в развитии нефросклероза объясняет попытки использовать их в качестве диагностических и прогностических маркеров ДН. Концентрация ММР-2 в крови и моче оказалась повышенной у больных СД1. Мочевая экскреция фермента показала взаимосвязь с наличием гиперфильтрации и микроальбуминурии [42]. Установлено, что признаком ДН при СД2 является повышенная активность ММР-9 в моче [41]. Повышение активности MMP-9 в плазме крови оказалось предиктором развития ДН у больных СД2 [43]. Наличие нефропатии у больных СД2 ассоциировано с повышенным уровнем тканевого ингибитора металлопротеиназ в крови [44].
Увеличение мочевой экскреции гликозаминогликанов (ГАГ) – возможный маркер ДН. Для оценки метаболизма протеогликанов в клинике изучают количество и состав ГАГ, экскретируемых с мочой. Предполагают, что в норме ГАГ попадают в мочу только путем фильтрации; секреции и реабсорбции этих веществ не установлено. Имеются данные, что гиперэкскреция ГАГ может указывать на скрытую дисфункцию почечного фильтра. У больных СД с нормальной ЭАМ стимуляционная проба с лизином выявляет патологическое увеличение альбуминурии [45]. Нами установлено, что суммарная экскреция сульфатированных ГАГ увеличивается уже в первые месяцы клинического течения СД1 и достигает наибольших значений у больных с микро- и макроальбуминурией, коррелируя с объемом мезангия [5].
В норме основным видом ГАГ, определяющимся в моче, считают хондроитинсульфат, в качестве минорной фракции может присутствовать гепарансульфат. Оказалось, что появление микроальбуминурии у больных СД1 связано со значительным увеличением экскреции гепарансульфата [5, 46]. Поскольку гепарансульфат-содержащие протеогликаны определяют отрицательный заряд базальной мембраны клубочков и препятствуют прохождению через почечный фильтр отрицательно заряженных молекул альбумина, можно предполагать, что “потеря” гепарансульфата с мочой способствует повышению ЭАМ.
Таким образом, лабораторные маркеры метаболизма внеклеточного матрикса у больных СД дают ценную информацию о развитии процессов склерозирования в почках и могут использоваться в целях диагностики и мониторинга течения нефропатии. При оценке данных маркеров следует учитывать наличие сопутствующих заболеваний (недиабетические поражения почек, болезни костей, диффузные болезни соединительной ткани и др.), которые могут быть причиной изменений обмена матрикса.
Факторы роста и цитокины. Определение медиаторов фиброгенеза представляет собой новый подход к диагностике и мониторингу течения прогрессирующих заболеваний почек.
Повышение мочевой экскреции трансформирующего фактора роста β (ТФР-β) зафиксировано у больных СД с микро- и макроальбуминурией [47–48]. Это согласуется с данными литературы о повышении почечной экспрессии гена ТФР-β при СД [49]. В настоящее время ТФР-β рассматривается как ключевой медиатор в формировании нефросклероза. Патогенетическая роль ТФР-β связана с активацией синтеза коллагена и других компонентов матрикса в почках [5, 50]. По нашим данным, увеличение мочевой экскреции ТФР-β у больных СД1 на допротеинурических стадиях ДН коррелирует с толщиной клубочковой и канальцевой базальной мембран и взаимосвязано с появлением коллагена I типа в клубочках [5, 48]. Терапия ингибиторами АПФ и антагонистами рецепторов ангиотензина II у больных СД сопровождается уменьшением мочевой экскреции ТФР-β, параллельным снижению протеинурии [51, 52].
У больных ДН наблюдается высокий уровень активной формы ТФР-β в сыворотке крови [53]. Установлено, что при ДН нарушена почечная регуляция уровня ТФР-β. Если в норме почки эффективно удаляют данный фактор из циркуляции, то у больных СД они, напротив, становятся его поставщиками [54].
Фиброгенный эффект ТФР-β в “диабетических” почках в значительной степени усиливается способностью этого медиатора активировать синтез фактора роста соединительной ткани (ФРСТ) – другого стимулятора продукции матрикса [55]. Экскреция ФРСТ с мочой резко возрастает у больных СД2 с микро- и макроальбуминурией, коррелируя с увеличением ЭАМ в динамике заболевания [56]. У больных СД1 с нефропатией повышение содержания ФРСТ в плазме крови коррелирует со снижением скорости клубочковой фильтрации и является предиктором развития уремии [57].
Инсулиноподобный фактор роста (ИФР-1) вовлечен в развитие ДН начиная с самых ранних стадий данного осложнения. Обсуждается роль ИФР-1 в развитии гипертрофии почек, клубочковой гиперфильтрации и гломерулосклероза [5, 58]. Мочевая экскреция ИФР-1 возрастает по мере развития ДН, начиная со стадии микроальбуминурии [59, 60]. У больных СД1 без протеинурии экскреция ИФР-1 коррелирует с толщиной базальной мембраны клубочков и канальцев [60].
В последние годы большое внимание отводят фактору роста эндотелия сосудов (ФРЭС). Большинство авторов рассматривают его как патогенный фактор в развитии ДН, поскольку ФРЭС способен увеличивать проницаемость клубочковых капилляров, синтез компонентов матрикса и пролиферацию мезангиальных клеток [5, 61]. Нами и другими исследователями зафиксировано повышение мочевой экскреции ФРЭС у больных СД с протеинурией [60, 62]. Это согласуется с данными о возрастании экспрессии ФРЭС в почках у пациентов с ДН [63]. Содержание ФРЭС в моче больных СД1 коррелирует с объемом мезангия клубочков [60].
Хотя традиционно ДН рассматривают как невоспалительное поражение почек, в последние годы установлено, что в развитии данного осложнения принимают участие воспалительные реакции [64, 65], следовательно, развитие ДН может отражать и признаки активации некоторых воспалительных цитокинов, а атакже хемокинов. Выявлена ассоциация ДН с воспалительными маркерами [66, 67]. Показана роль мигрирующих в почки воспалительных клеток, прежде всего моноцитов/макрофагов, в развитии гломерулярного и интерстициального фиброза при СД [68, 69]. Установлено, что миграция мононуклеаров и реализация их активности осуществляются в тесном взаимодействии с резидентными клетками и внеклеточным матриксом почек. Важнейшую роль в этом взаимодействии играют провоспалительные цитокины, в частности интерлейкин-1β (IL-1β), моноцитарный хемоаттрактантный протеин-1 (МСР-1), хемокин, экспрессируемый и секретируемый Т-клетками при активации (RANTES) [65, 70].
Проведенное нами исследование показало повышение экскреции провоспалительных цитокинов у больных СД1: экскреция IL-1β была повышена при любой ЭАМ, экскреция MCP-1 – у больных с микро- и макроальбуминурией, экскреция RANTES – у больных с макроальбуминурией. У пациентов с допротеинурическими стадиями ДН экскреция цитокинов коррелировала с толщиной базальной мембраны клубочков и канальцев, при этом наиболее тесную взаимосвязь показал IL-1β [60]. Другие исследователи также зафиксировали повышение мочевой экскреции МСР-1 по мере развития ДН [71, 72]. Данные о гиперэкскреции IL-1β и RANTES получены нами впервые. Усиление экспрессии MCP-1 и RANTES в клубочках, в большей степени – в канальцах почек, обнаружено у больных СД с нефропатией. Показано, что активация транскрипции генов цитокинов в канальцах при ДН связана с воспалительной инфильтрацией интерстиция и степенью протеинурии [73, 74]. Повышение экскреции МСР-1 у больных СД2 с нефропатией оказалось прогностическим маркером снижения фильтрационной функции почек [55].
Таким образом, определение циркулирующих, особенно экскретируемых, фиброгенных факторов роста и провоспалительных цитокинов может использоваться для оценки процессов фиброгенеза и хронического воспаления в почках при СД.
В последние годы появились сообщения об увеличении мочевой экскреции ангиотензиногена [75], нефрина [76]; изменении экспрессии белков подоцитов в клеточном осадке мочи [77] у больных СД с различными стадиями нефропатии. Диагностическое и прогностическое значения этих маркеров нуждаются в уточнении. Новые перспективы для ранней диагностики ДН открывает протеомика мочи [78, 79].
Таким образом, в настоящее время стандартом обследования больных СД является определение альбуминурии. Микроальбуминурия является наиболее ранним маркером ДН, выявляемым в рутинной клинической практике. Проведенные к настоящему времени исследования доказали, что альбуминурия коррелирует с выраженностью морфологических изменений в почках, однако начальные структурные признаки ДН появляются при нормальной ЭАМ. Альбуминурия является достаточно вариабельной величиной, ее значимость как предиктора прогрессирования ДН далеко не абсолютна. В связи с этим актуальной задачей остается поиск других диагностических и прогностических маркеров диабетического нефросклероза. Перспективным направлением такого поиска является изучение маркеров обмена компонентов внеклеточного матрикса, факторов роста, цитокинов и других молекул, вовлеченных в развитие ДН.



{kind=link}
{kind=link}