
Введение
При уремии возникает необходимость резкого снижения содержания азота в питании при сохранении удовлетворения потребности человеческого организма в незаменимых аминокислотах. Для этого последние заменяют в диете соответствующими α-кетокислотами или находящимися с ними в равновесии α-гидроксикислотами в расчете на то, что при переаминировании кетокислоты превратятся в аминокислоты: α-кетоаналоги лейцина, изолейцина, валина, фенилаланина (рис. 1) и α-гидроксианалог метионина. В то же время такие незаменимые аминокислоты, как лизин, треонин, триптофан, а также становящиеся при уремии незаменимыми (условно незаменимыми) гистидин и тирозин вводят в организм в виде аминокислот [1].
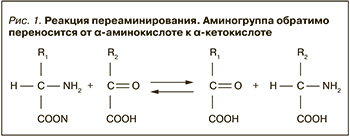
В своей монографии в 1969 г. Т.Т. Березов писал: считается доказанным, что в тканях млекопитающих механизм переаминирования – главный путь дезаминирования L-изомеров фенилаланина, тирозина, лейцина, изолейцина, валина, орнитина, цистеиновой и цистеинсульфиновой кислот, β-аланина, γ-аминомасляной кислоты и частично цистеина. Тот же механизм должен быть отнесен к дезаминированию L-триптофана, которому предшествует разрыв индольного ядра с образованием кинуренина и далее 3-оксикинуренина, которые или подвергаются прямому переаминированию с α-кетоглутаровой кислотой, или распадаются под действием специфической кинурениназы с образованием аланина. Дезаминирование аланина также происходит путем переаминирования. Имеются данные о преимущественной роли переаминирования в дезаминировании аланина, аспарагиновой кислоты, метионина, треонина, серина и др. в печени и почках [2]. При этом Т.Т. Березов не называет лизин, следовательно, он имеет в виду непсредственно аминокислоты, а не продукты их превращения, потому что продукт превращения лизина – α-аминоадипиновая кислота – активно подвергается переаминированию [3]. Т.Т. Березов также отмечает, что переаминирование может происходить в тканях между разнообразными монокарбоновыми донорами и акцепторами аминогрупп без участия дикарбоновых аминокислот [2]. К реакциям этого типа относятся процессы переаминирования между рядом аминокислот и пировиноградной кислотой с образованием аланина и соответствующих α-кетокислот, протекающие в митохондриях печени. Показана и обратимость этих реакций (рис. 1), а также различная способность отдельных тканей катализировать описанные превращения.
Млекопитающим, больным уремией и находящимся на низкобелковой диете, скармливали α-кетокислоты – производные незаменимых аминокислот [4]. Последние синтезировались в организме путем переаминирования. Оказалось, что в виде аминокислот необходимо давать только лизин, для которого отсутствует трансаминаза [5]. В то же время степень использования α-кетокислот для синтеза соответствующих аминокислот различается. Валин, лейцин, изолейцин, метионин и фенилаланин быстро синтезировались путем переаминирования, в то время как гистидин (для подавляющего числа млекопитающих он считается незаменимой аминокислотой, а для здоровых взрослых людей – заменимой), треонин и триптофан синтезировались в меньшей степени, а синтеза лизина вообще не наблюдалось.
В то же время выше уже говорилось, что в питании людей, больных уремией, некоторые аминокислоты не замещают α-кето- или гидроксианалогами. Однако, чтобы сделать вывод, какие незаменимые аминокислоты могут быть заменены в диете этими аналогами, а какие нет, необходимо рассмотреть изменения превращения каждой незаменимой аминокислоты при уремии. Насколько мне известно, такая работа не проводилась. В частности, треонин не заменяется безазотистыми аналогами, исходя из сложившегося стереотипа, что он, как и лизин, не подвергается переаминированию. Между тем переаминирование треонина показано, как уже отмечалось, у млекопитающих, больных уремией [4] и у здоровых людей [3]. Может ли замещение треонина его безазотистыми аналогами в диете людей, больных уремией, компенсировать потребность в нем, сумеет показать только практическое применение этих аналогов. Во всяком случае ничего не известно об усилении распада треонина при уремии, как это имеет место в отношении циклических аминокислот, кроме фенилаланина.
Вторым вопросом, поднятым в статье, будет необходимость обязательной добавки витамина В12 к гидроксианалогу метионина. Если метионин будет синтезироваться путем переаминирования из своего кетоаналога, в который окислится гидроксианалог, это не значит, что основная доля метионина будет распадаться путем переаминирования: она будет, как и в норме, распадаться путем переметилирования, а чтобы последнее оставалось при уремии обратимым, необходимо дополнительное введение в организм витамина В12, который, став коферментом, катализирует реакцию метилирования гомоцистеина в метионин.
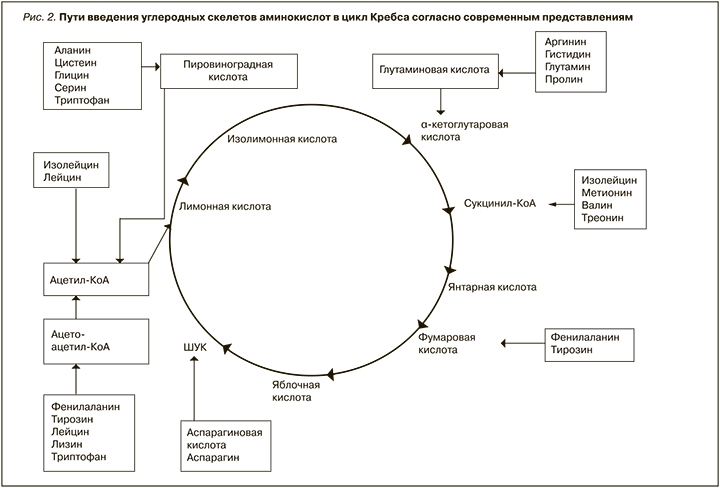
Третий вопрос, поднятый в статье, следующий. Все присутствовавшие в диете α-кето- и гидроксианалоги – безазотистые вещества, а потому соответствуют критерию снижения азота в питании. Однако нельзя забывать, что некоторые нефропатии, при которых распад циклических аминокислот становится столь интенсивным, что замещение кетоаналогами не может компенсировать потребность организма в этих аминокислотах, являются осложнениями сахарного диабета (СД) [6–9], при котором особенно ярко проявляется глюкогенный или кетогенный характер той или иной аминокислоты, имеющий место и в здоровом организме, обусловленный спецификой превращения различных аминокислот (рис. 2) [10]. Введение в организм кетогенных аминокислот отягчает проявление имеющегося при СД кетоза, в то время как введение глюкогенных аминокислот снижает тяжесть последнего в силу их антикетогенного действия. Последнее обусловлено тем, что раньше, чем в глюкозу, глюкогенные аминокислоты превращаются в тот или иной компонент цикла Кребса или в пировиноградную кислоту, находящуюся в равновесии с этими компонентами, что стимулирует окисление ацетил-КоА, а следовательно, кетоновых тел. Сент-Дьерди называл это сгоранием жиров в пламени углеводов, а кетоновые тела как раз служат промежуточным продуктом жирового обмена. Все присутствующие в диете безазотистые аналоги незаменимых аминокислот обладают тем же глюкогенным либо кетогенным действием, что и последние. Кроме того, аминокислоты, присутствующие в диете в неизменном виде, также обладают глюкогенным либо кетогенным действием. Этот факт нигде прежде не рассматривался, а он должен обязательн учитываться в случае диабетического происхождения нефропатии. Кетоз при СД возникает по двум причинам: а) если у здорового человека основным источником энергии являются углеводы, то при СД таковым становятся жиры, которые интенсивно окисляясь, образуют повышенное количество кетоновых тел, б) снижение образования щавелевоуксусной кислоты (ЩУК) из пировиноградной кислоты в силу снижения образования последней из глюкозы и увеличения использования компонентов цикла Кребса для глюконеогенеза. При кетозе происходит: 1) резкий сдвиг рН в кислую сторону вследствие накопления в крови ацетоуксусной и β-оксимасляной кислот, 2) наркотическое действие третьего кетонового тела-ацетона. Диабетическая кома как раз служит следствием кетоза. В большинстве случаев причиной летального исхода СД считается диабетическая кома, к которой приводит декомпенсированная форма кетоза. Следует также добавить, что причиной смерти от диабетической комы является не наркотическое действие ацетона, а сдвиг рН в кислую сторону, что нарушает работу ферментов.
Цели обзора:
1) биохимическое обоснование замещения или его отсутствия α-кето- и гидроксианалогами в диете больных уремией каждой незаменимой аминокислоты, а также двух заменимых для здоровых людей аминокислот, но становящихся для больных уремией условно-незаменимыми; предложение замещения треонина его безазотистыми аналогами;
2) биохимическое обоснование введения в организм витамина В12 вместе с безазотистым аналогом метионина с целью уменьшения расхода метионина в организме и возможностью компенсации потребности в метионине заменой его безазотистым аналогом;
3) требование установления верхнего предела введения в организм кетогенных аминокислот и их α-кето- и гидроксианалогов при диабетическом происхождении нефропатии.
Незаменимые аминокислоты
1. Аминокислоты с разветвленной углеродной цепью
К аминокислотам с разветвленной углеродной цепью относятся три незаменимые аминокислоты: лейцин, изолейцин и валин. Все они легко подвергаются обратимому переаминированию с дальнейшим необратимым окислением α-кетокислот в отсутствие каких-либо еще путей распада в организме человека, поэтому в диете больных уремией заменяются соответствующими α-кетокислотами.
В то же время валин имеет только глюкогенное действие, лейцин – только кетогенное, изолейцин – глюкогенное и кетогенное одновременно. Все это относится и к их кетоаналогам. Поэтому, если в анамнезе нефропатии лежит СД, не может стоять вопроса об ограничении в диете валина, чего нельзя сказать о лейцине и изолейцине, введение которых в организм может усиливать имеющийся при СД кетоз.
В данном случае необходимо подобрать оптимальное количество кетоаналогов этих двух аминокислот, которое, с одной стороны, удовлетворяло бы потребность организма в лейцине и изолейцине, с другой – не усиливало бы проявления кетоза.
2. Метионин
Единственная серосодержащая незаменимая аминокислота. Она так же легко подвергается обратимому переаминированию, поэтому так же замещается в питании больных уремией безазотистым аналогом, только при этом используют не α-кетокислоту в силу ее неустойчивости, а α-гидроксикислоту. Однако распад метионина в организме осуществляется не путем переаминирования, а путем переметилирования, превращающего метионин в гомоцистеин (аминокислоту, не входящую в состав белков) с дальнейшим ее необратимым распадом либо реметелированием в метионин, основная доля которого осуществляется при обязательном участии витамина В12 в качестве кофермента, что также необходимо учитывать с целью уменьшения в организме расхода метионина при уремии. Дело в том, что превращение метионина в гомоцистеин и обратный процесс катализируются совершенно различными ферментами. Метильная группа метионина необратимо теряется при синтезе креатина, карнитина, адреналина, метилникотинамида и др., и для этих реакций витамин В12 не требуется. Для биосинтеза же одной молекулы холина, содержащей три метильные группы, требуется три молекулы метионина. Когда холин окисляется в бетаин, последний переметилируется с гомоцистеином
с возращением 1 метильной группы метионину также без участия витамина В12, но этого явно недостаточно для восстановления концентрации метионина. В то же время в организме есть фермент, катализирующий необратимое метилирование гомоцистеина в метионин, и его коферментом является витамин В12. Поэтому необходимой добавкой к безазотистому аналогу метионина с целью обеспечения организма этой аминокислотой является витамин В12. Интересно отметить, что именно эта реакция лежит в основе кроветворного действия витамина В12, т.к. способствует освобождению тетрагидрофолиевой кислоты от балластной метильной группы для участия тетрагидрофолиевой кислоты в синтезе пуриновых и пиримидиновых оснований.
Метионин обладает только глюкогенным действием, поэтому не может представлять опасность при диабетических нефропатиях (ДН).
3. Лизин
По последним данным, из всех природных аминокислот только лизин не способен подвергаться переаминированию [4, 5], поэтому может применяться только в виде аминокислоты. Лизин распадается в животном организме исключительно необратимо начиная с прямого окислительного дезаминирования.
Лизин обладает только кетогенным действием, что должно учитываться при ДН.
4. Циклические аминокислоты
Из четырех циклических аминокислот для здорового взрослого человека незаменимы две: триптофан и фенилаланин. Обе обладают одновременно глюкогенным и кетогенным действиями. Последнее должно учитываться при ДН.
Фенилаланин может так же активно подвергаться обратимому переаминированию, как аминокислоты с разветвленной углеродной цепью и метионин, в то время как триптофан – в значительно меньшей степени [4]. Но главный путь распада этих аминокислот сводится к необратимому окислению фенилаланина в тирозин, а триптофана – в кинуренин с последующим распадом этих соединений.
Поскольку при хронической болезни почек (ХБН) главной мишенью среди аминокислот становятся циклические аминокислоты в силу особенности их строения, на них стоит остановиться.
Фенилаланин является ароматической аминокислотой. Его молекула достаточно устойчива даже при развивающемся при уремии оксидативном стрессе. Кроме того, отмечено резкое повышение при уремии в плазме крови отношения фенилаланин/тирозин, причем за счет снижения концентрации тирозина; концентрация фенилаланина оставалась нормальной или несколько повышенной (в дальнейшем мы еще к этой теме вернемся) [11]. Следовательно, больные уремией не испытывают дефицита фенилаланина и его замена в диете соответствующей α-кетокислотой полностью компенсирует потребность в фенилаланине.
Что касается триптофана, то здесь картина противоположная. Будучи не ароматической, а гетероциклической аминокислотой, триптофан имеет молекулу, которая при уремии становится неустойчивой.
Отмечено, что у всех пациентов с почечной недостаточностью до лечения гемодиализом резко падал уровень триптофана в сыворотке крови по сравнению со здоровыми субъектами и при этом резко увеличивался уровень продуктов окисления триптофана: кинуренина и хинолиновой кислоты [12].
Отношение кинуренин/триптофан в плазме крови считают чувствительным и удобным методом определения функции почек, т.к. при уремии оно достигало 103%, причем за счет как резкого снижения концентрации триптофана, так и увеличения концентрации кинуренина [13]. Растет также концентрация образующейся из кинуренина кинуреновой кислоты. Установлено, что эти изменения обусловлены возрастанием активности ферментов, катализирующих необратимое окисление триптофана в кинуренин.
Появление усталости при хронической болезни почек (ХБП) объясняют снижением образования мелатонина вследствие сниженного уровня триптофана в организме [14]. Снижение уровня триптофана обусловлено повышением активности фермента, необратимо окисляющего триптофан, в результате воспаления, развивающегося при ХБП.
В исследовании патогенеза воспаления у больных уремией, вызываемого в т.ч. продуктами распада триптофана, констатировали, что воспаление повышает активность ферментов, окисляющих триптофан в кинуренин [15].
S. Debnath et al. впервые исследовали метаболизм триптофана и маркеров воспаления у больных СД и ХБП различных стадий, включая терминальную [8]. Результаты показывают заметное уменьшение циркуляции триптофана пропорционально уменьшению функции почек. По сравнению со стадией 1 ХБП уровни триптофана плазмы крови были на 60% ниже на стадии 5 ХБП. Уменьшение триптофана сопровождалось увеличением уровней кинуренина и кинуреновой и хинолиновой кислот: это предварительное наблюдение обнаружило индукцию ферментов распада триптофана. Циркуляционные уровни метаболитов триптофана положительно и прочно коррелировали не только с выраженностью почечной недостаточности, но и с цитокинами, вызывающими воспаление.
C.-A. Chou et al. впервые используют концентрацию триптофана в сыворотке крови при СД как прогностический фактор в развитии почечной недостаточности [9]. Низкий уровень триптофана служит маркером ДН. Авторы считают, что такое уменьшение концентрации триптофана объясняется хлорированием и оксидированием остатков триптофана в коллагене, причем последнее усугубляет почечную недостаточность. Таким образом, не только почечная недостаточность приводит к усилению ферментативного распада триптофана, но и возникающий при СД оксидативный стресс приводит к неферментативному окислению остатков триптофана в составе белков, что усиливает почечную недостаточность.
V.C. Konje et al. впервые исследовали связь кинуренинового пути распада триптофана с сердечно-сосудистой недостаточностью, развивающейся у больных уремией [16]. Открытия наводят на мысль о роли низких уровней триптофана в плазме крови уже на ранних стадиях сердечно-сосудистой недостаточности в популяции больных ХБП.
Y. Cheng et al. исследовали связь метаболитов триптофана с функцией почек и констатировали, что при снижении функции почек уровень триптофана в крови снижается в отличие от кинуренина [17]. Это могло бы означать возросшую активность ферментов, катализирующих превращение триптофана в кинуренин [12].
I. Kwiatkowska et al. отмечают, что, несмотря на то что в норме триптофан метаболизируется главным образом в печени, при ХБП его превращение резко изменяется [18]: происходит уменьшение концентрации триптофана в сыворотке крови больных и при этом увеличивается отношение кинуренина к триптофану, что показывает повышенную активность ферментов, расщепляющих триптофан [15]. В работе анализируются причины повышения активности этих ферментов на гуморальном уровне (эти сведения выходят за рамки темы данной статьи). Причем почки вносят вклад в эти изменения двумя путями. Во-первых, они характеризуются высокой экспрессией ферментов (название их выходит за рамки данной статьи), усиливающих распад триптофана. Во-вторых, по мере развития дисфункции почек выведение с мочой продуктов распада триптофана становится недостаточным, что способствует накоплению этих продуктов, а это может провоцировать оксидативный стресс и как следствие – распад триптофана [9].
Итак, ХБП характеризуется резким снижением в организме количества триптофана. У млекопитающих он способен синтезироваться из своего кетоаналога путем переаминирования [4], но даже в норме 95% триптофана необратимо распадается по кинурениновому пути [19, 20], а из оставшихся 5% только часть подвергается переаминированию. У здорового человека эта часть еще меньше, чем у млекопитающих, за счет большего превращения триптофана в серотонин в мозге. Все это делает невозможным замещение триптофана соответствующей α-кетокислотой в диете больных уремией и предписывает им потреблять триптофан в неизменном виде.
5. Треонин
Треонин является β-гидроксиаминокислотой, как и заменимая аминокислота серин, поэтому некоторые реакции превращения в организме совпадают у этих двух аминокислот. Так, единственной в организме человека реакцией распада углеродного скелета треонина остается необратимая дегидратация, которой подвергается и серин под влиянием того же фермента. Дальнейшее превращение этих двух аминокислот обусловливает их глюкогенное действие в отсутствие кетогенного [21]. Но серин может синтезироваться в организме различными путями, в т.ч. переаминированием своего кетоаналога, который легко образуется в организме из глюкозы. Кетоаналог треонина подобно кетоаналогам других незаменимых аминокислот не синтезируется в организме человека и животных. И здесь возникает вопрос: может ли треонин в диете больных уремией заменяться соответствующей α-кетокислотой?
В работе A.P. Shah et al. повторяется стереотипное суждение, согласно которому треонин, как и лизин, не способен к обратимому переаминированию, а потому не может быть замещен в диете соответствующей α-кетокислотой [1]. Между тем выше уже отмечалось, что синтез треонина из его кетоаналога у млекопитающих, больных уремией, происходит путем переаминирования в степени, соизмеримой с триптофаном и гистидином [4].
Переаминирование треонина показано и у человека [3, 22, 23]. Из его печени выделены трансаминазы широкого профиля, катализирующие переаминирование многих аминокислот, включая треонин [22, 23]. Из мозга человека выделена трансаминаза, способная переаминировать 16 аминокислот с 16 α-кетокислотами [23]. Среди первых называется и треонин, но среди вторых его кетоаналог не называется. Из этого можно сделать следующий вывод: α-кето-β-гидроксимасляная кислота, будучи химически неустойчивым соединением, быстро (но обратимо) восстанавливается в α-, β-дигидроксимасляную кислоту с помощью НАДН или НАДФН и соответствующей дегидрогеназы, а потому в организме не обнаруживается.
Выше уже говорилось и ниже будет показано резкое возрастание неустойчивости при уремии ряда циклических аминокислот, что делает невозможным их замещение в диете соответствующими кетоаналогами. Но треонин – ациклическая аминокислота. Исходя из этого, возникает возможность замещения треонина в питании больных уремией тем или иным безазотистым аналогом: α-кето-β-гидроксимасляной кислотой, а в случае ее неустойчивости – α-, β-дигидроксимасляной кислотой (подобно тому, как метионин заменяют его гидроксианалогом). Будет ли при этом удовлетворено обеспечение организма треонином, может показать только практическое использование того или иного безазотистого аналога треонина.
Выше также отмечалось: распадаясь в организме человека путем дегидратации, треонин проявляет только глюкогенное действие. Что касается его α-, кето- и гидроксианалогов, то они в процессе распада превращаются в молочную кислоту и потому, подобно треонину, обладают только глюкогенным действием.
Условно-незаменимые аминокислоты
К условно-незаменимым аминокислотам относятся две ци-клические аминокислоты: тирозин и гистидин, заменимые для здорового человека, но становятся незаменимыми при уремии.
Тирозин – заменимая аминокислота для здоровых людей и животных, т.к. является нормальным продуктом превращения незаменимой амнокислоты фенилаланина (если у человека с рождения этого превращения не происходит, развивается фенилпировиноградная олигофрения). Основным путем распада тирозина в организме является обратимое переаминирование с дальнейшим необратимым окислением α-кетокислоты с разрывом бензольного кольца. Легко догадаться, что, как и его предшественник, фенилаланин, тирозин одновременно обладает глюкогенным и кетогенным действиями.
Выше уже говорилось, что в плазме крови при уремии отмечено резкое повышение отношения фенилаланин/тирозин за счет снижения концентрации тирозина [11]. В состав белков входят только две ароматические аминокислоты: фенилаланин и его гидроксипроизводное тирозин. И здесь проявляются их противоположные химические свойства: если при оксидативном стрессе, вызванном уремией, фениллаланин остается устойчивым (см. выше), то тирозин проявляет крайнюю неустойчивость. Так, H. Ischiropoulos указывает, что при оксидативном стрессе по мере развития его тяжести происходит биологический процесс нитрования тирозина путем биохимического взаимодействия с окисью азота (NO) или образующихся из нее вторичных продуктов [24]. Этот процесс происходит почти во вcех органах, причем сильно сказывается на их функции, т.к. при этом происходит нитрование остатков тирозина в белках этих органов. R.C. Thuraisingham et al. предполагают, что гипергликемия, являющаяся главным симптомом СД, вызывает повышенное образование радикалов окиси азота, которые взаимодействуют с остатками тирозина в белках почечной ткани, превращая тирозин а нитротирозин, что и приводит к почечной недостаточности [6]. G. Deng et al. говорят о значении избыточного нитрования тирозина в коре головного мозга при уремии в патогенезе уремической энцефалопатии и снижение тяжести последней при антиоксидантной терапии [25]. Все это указывает на роль оксидативного стресса в усилении нитрования тирозина в мозге при уремии.
Но не только усиление процесса нитрования тирозина ответственно за резкое снижение количества последнего у больных уремией. До этого речь шла о распаде тирозина, а теперь стоит поговорить о его образовании из фенилаланина.
P. Tessari et al. отмечают, что гидроксилирование в тирозин – это главный путь превращения фенилаланина в почках [26]. Причем если в почках человека гидроксилирование фенилаланина в тирозин насчитывает 70% фенилаланина, то в органах брюшной полости только 8%. Сделанные в этой работе открытия важны для понимания как почечного метаболизма в физиологических условиях, так и изменений азотистого обмена при уремии. В частности, эти открытия объясняют имеющий место при уремии сниженный пул тирозина. U. Lichter-Konecki et al. сообщают о выделенном из почек человека гене фенилаланиндегидрогеназы, что говорит о заметной роли почек в гомеостазе фенилаланина, проявляющейся в наблюдаемом при почечной недостаточности сниженном гидроксилировании фенилаланина в тирозин [27]. N. Moller et al. провели исследования, в которых показали, что превращение фенилаланина в тирозин происходит в количестве 5,2±1,2 мкмоль/мин в почках и 3,0±0,7 мкмоль/мин в органах брюшной полости, включая печень [28]. Почки вносят вклад в существенное количество тирозина в общем кровотоке, в то время как в органах брюшной полости тирозин распадается до конечных продуктов и не поступает в общий кровоток. Эти результаты демонстрируют, что почки – главный донор тирозина общего кровотока благодаря окислению в них фенилаланина в тирозин. Поэтому, хотя превращение фенилаланина в тирозин имеет место как в почках, так и в органах брюшной полости, только почки обеспечивают тирозином общий кровоток и тем самым другие ткани для синтеза белков и ряда небелковых азотистых физиологически активных веществ. Y. Boine et al. отмечают, что по этой причине в терминальной стадии почечной недостаточности степень превращения фенилаланина в тирозин на 50% ниже, чем у здоровых людей [29]. Таким образом, почки обеспечивают по крайней мере 50% превращения в организме фенилаланина в тирозин. В работе тирозин считают незаменимой аминокислотой для этих больных в силу нарушения его образования из фенилаланина.
J.D. Kopple et al. показали, что при ХБП нарушается гидроксилирование и удаление фенилаланина, уменьшается синтез тирозина из фенилаланина и накапливаются метаболиты фенилаланина и тирозина из-за снижения их экскреции с мочой [7]. При ХБП также имеются другие нарушения в биохимии этих аминокислот. Индивидуумы с ХБП часто подвержены оксидативному стрессу, который приводит к увеличенному образованию или уменьшенному устранению продуктов окисления, свободных радикалов, хлорноватистой кислоты и NO2. Эти соединения реагируют с тирозином, приводя к образованию 3-хлортирозина и нитротирозина в белках плазмы крови. Нитротирозин также увеличивается в почках больных ДН [6]. Кроме того, увеличивается содержание нитротирозина в белках мозга больных ХБП [25].
Итак, тирозин, как и триптофан-циклическая аминокислота, при оксидативном стрессе подвергается, подобно триптофану, разрушению как в свободном виде, так и в составе белков. Так что, несмотря на то что тирозин легко может синтезироваться путем переаминирования из соответствующей α-кетокислоты [4], прием последней не может обеспечивать тирозином организм больного уремией. А посему его надлежит вводить в организм в неизменном виде.
Особенность гистидина в следующем: будучи незаменимой аминокислотой для подавляющего количества животных, она заменима для здоровых взрослых людей [30]. Здесь необходимо отметить такую особенность гистидина: если все другие аминокислоты в животном организме подвергаются только окислительному дезаминированию (прямому или через переаминирование), при этом превращаясь в соответствующие α-кетокислоты, то гистидин подвергается прямому необратимому дезаминированию, превращаясь в соответствующую непредельную кислоту. Дальнейший ее необратимый распад приводит к разрыву имидазольного кольца и образованию глутаминовой кислоты, обладающей глюкогенным действием. Следовательно, гистидин обладает только глюкогенным действием. В значительно меньшей степени гистидин подвергается обратимому переаминированию [4], но та часть гистидина, которая подвергается последнему, также претерпевает дальнейший разрыв имидазольного кольца с образованием аспарагиновой кислоты, обладающей глюкогенным действием.
Будучи, как и триптофан, гетероциклической аминокислотой, гистидин при уремии проявляет крайнюю неустойчивость молекулы.
В исследовании больных уремией и одного здорового добровольца выявлено, что больные уремией делят неспособность синтезировать гистидин со всеми животными, изученными в этом отношении, а также с новорожденными детьми [31]. В работе отмечается снижение концентрации гистидина в плазме крови при уремии. F.B. Stifel et al. говорят, что определенные болезненные состояния могут быть охарактеризованы неспособностью синтезировать гистидин или увеличением его распада [32]. M. Watanabe et al. на основе недавних исследований определяют гистидин как заменимую аминокислоту для здоровых взрослых людей и как условно-незаменимую – для больных уремией [33]. В работе исследуется связь гистидина плазмы крови как противовоспалительного и антиоксидантного фактора с маркерами воспаления и оксидативного стресса и фактором роста гепатоцитов у больных уремией. Авторы пишут, что причины низких внеклеточной и внутриклеточной концентраций гистидина при уремии неясны, однако предполагают, что это связано с воспалением и оксидативным стрессом, имеющими место при уремии. Введение в организм больных уремией гистидина не только восстанавливает азотистое равновесие, но и оказывает противовоспалительное и антиоксидантное действия [34].
Естественно, потребность организма в гистидине при уремии может быть удовлетворена только путем введения в организм этой аминокислоты в неизменном виде.
Следует также учесть, что небольшая часть вводимых в организм больных уремией триптофана и тирозина в процессе распада подвергается декарбоксилированию, превращаясь в тканях в амины триптамин и тирамин соответственно. Кроме того, триптофан в мозге может окисляться в 5-гидрокситриптофан, превращающийся при декарбоксилировании в амин серотонин, а тирозин – в 3,4-диоксифенилаланин (ДОФА), декарбоксилирующийся в амин дофамин, который затем может окисляться в норадреналин. Все перечисленные амины считаются физиологически активными веществами, а потому существует настоятельная необходимость изучения изменения превращения в них данных аминокислот при уремии. В то же время дальнейшее окисление в организме этих аминов не приводит к разрыву колец в их молекулах. Поэтому распад тирозина и триптофана по данному пути не может иметь ни глюкогенное, ни кетогенное действия. О превращениях аминокислот в гормоны (тирозина – в адреналин и тироксин и гистидина – в гистамин) говорить не приходится.
Итак, представленные данные по каждой аминокислоте говорят: аминокислоты вполне обоснованно заменяются или не заменяются в диете больных уремией α-, кето- и гидроксианалогами, исключение составляет лишь треонин. Насколько замена последнего в диете безазотистыми аналогами может обеспечить в нем организм, может показать только практическое их применение. Для обеспечения же организма метионином его безазотистый аналог необходимо вводить в организм вместе с витамином В12 . При диабетической уремии необходимо также учитывать, глюкогенным или кетогенным действием обладает та или иная аминокислота либо ее аналог, и в случае кетогенного действия должен быть установлен верхний предел введения аминокислоты в организм во избежание усиления кетоза.
Заключение
В статье рассматривается возможность замещения α-, кето- и гидроксианалогами в диете больных уремией 10 незаменимых для больных ХБП аминокислот, из которых две заменимы для здоровых людей. Анализируется основание замещения каждой аминокислоты ее аналогом. Со всеми применяемыми в настоящее время замещениями аминокислот их аналогами равно, как и с невозможностью этого замещения, следует согласиться, кроме невозможности замещения треонина. В организме человека треонин подвергается обратимому переаминироваиию вопреки устоявшемуся мнению. Это позволяло бы в диете больных уремией его замещать соответствующей α-кетокислотой, как это принято в отношении других незаменимых аминокислот: лейцина, изолейцина, валина и фенилаланина. Однако α-кетоаналог треонина α-кето-β-оксимасляная кислота не обнаружена в организме человека [23]. Весьма высока вероятность того, что она, будучи химически неустойчивым соединением, восстанавливается в α-, β-дигидроксимасляную кислоту. Поскольку это восстановление обратимо, последняя может замещать треонин в диете больных нефропатиями подобно тому, как метионин замещают его α-гидроксианалогом. Здесь необходимо добавить, что такие аминокислоты, как триптофан, тирозин и гистидин, также не заменяют в диете больных нефропатиями их α-, кето- или гидроксианалогами, а вводят в организм в виде аминокислот [29]. Все они являются циклическими аминокислотами, и их расход при нефропатиях резко усиливается, так что тирозин и гистидин, будучи заменимыми аминокислотами для здоровых людей, становятся условно-незаменимыми для больных нефропатиями. Триптофан считается незаменимой аминокислотой и для здоровых лиц (из циклических аминокислот только фенилаланин заменяют кетоаналогом). Иными словами, расход циклических аминокислот, кроме фенилаланина, при нефропатиях таков, что переаминирование их кетоаналогов не компенсирует потребности в этих аминокислотах. Треонин же является ациклической аминокислотой, и ничего неизвестно об увеличении его расхода при уремии, что вселяет надежду на его замену в диете безазотистыми аналогами.
Кроме того, потребность организма в метионине путем его замещения в диете безазотистым аналогом (в данном случае α-гидороксикислотой) может быть удовлетворена только в случае умеренного распада этой аминокислоты. Основным путем распада в организме метионина является переметилирование с образованием гомоцистеина, для чего витамина В12 не требуется, но для реметилирования гомоцистеина в метионин – требуется (в статье это объясняется). Поэтому возможность компенсации потребности в метионине заменой его в диете безазотистым аналогом реальна только при введении с последним в организм витамина В12, о чем также говорится в статье. Это введение может одновременно оказывать положительное действие при развивающейся при ХБП анемии.
Наконец, поскольку нефропатии могут иметь диабетическое происхождение, каждая аминокислота описывается в статье как имеющая глюкогенное либо кетогенное действие. Такие аминокислоты, как лейцин, изолейцин, лизин, триптофан, фенилаланин, тирозин, а также α-кетоаналоги лейцина, изолейцина и фенилаланина обладают кетогенным действием. Их введение в организм больного СД может усугубить тяжесть кетоза, имеющего место при этой болезни. Поэтому при ДН должен быть установлен четкий верхний предел введения в организм этих веществ.


