
Среди механизмов, определяющих течение и исход заболеваний почек, наиболее изученными в настоящее время являются повреждающие (эффекторные) звенья патогенеза – формирование клеточного воспалительного инфильтрата, высвобождение воспалительных цитокинов, метаболитов
арахидоновой кислоты и кислородных радикалов, активация компонентов комплемента. Однако степень повреждения ткани почки зависит от баланса локально воздействующих повреждающих факторов и противостоящих им медиаторов самозащиты, при этом активация факторов локальной самозащиты тканей в ответ на повреждение представляет собой универсальную запрограммированную реакцию.
Находясь в воспалительном микроокружении, клетки почечной ткани могут приобретать толерантность к воспалительным стимулам – потенциал самозащиты от дальнейшего повреждения [1]. Эндогенные протективные медиаторы, которые могут изменять течение патологического воспалительного процесса в почке, представляют особый интерес для понимания основных закономерностей прогрессирования поражения почек. В основе формирования “противовоспалительного статуса” ткани почки лежит активация факторов самозащиты на
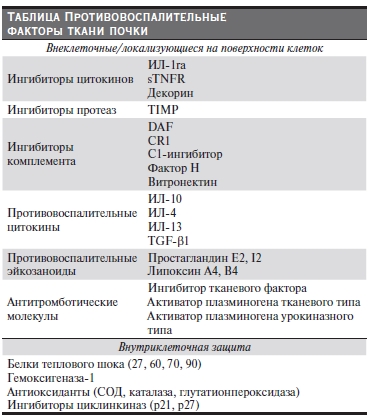
различных уровнях – вне- и внутриклеточном, а также на поверхности клеток (см. таблицу). В обзоре представлены протективные факторы, введение которых экспериментальным животным эффективно подавляет воспаление при различных иммуновоспалительных заболеваниях и является перспективным для разработки новых направлений терапевтического воздействия на человека.
Ингибиторы воспалительных медиаторов
Одним из механизмов ограничения воспаления в почке является выделение специфических ингибиторов провоспалительных медиаторов, которые могут секретироваться как клетками воспалительного инфильтрата, так и резидентными клетками ткани почки.
Антагонист рецептора интерлейкина-1 (ИЛ-1ra)
Интерлейкин-1 (ИЛ-1) – провоспалительный цитокин, продуцируется клетками воспаления, вызывает высвобождение и экспрессию других воспалительных медиаторов (цитокинов/факторов роста, хемокинов, биоактивных липидов, металло-протеиназ и реактивных радикалов кислорода, адгезивных
рецепторов), пролиферацию резидентных клеток, накопление экстрацеллюлярного матрикса [2]. Естественным ингибитором ИЛ-1 является антагонист рецептора ИЛ-1 (ИЛ-1ra) [3]. В здоровых почках крыс экспрессия ИЛ-1ra не определяется, однако она существенно увеличивается при экспериментальном анти-БМК-нефрите. В этой модели экспрессия ИЛ-1ra достигает пика (10–20-кратного повышения) через 6 часов после индукции и персистирует до 4 суток. Но этого количества эндогенного ИЛ-1ra оказывается недостаточно для подавления ИЛ-1-индуцированного воспаления, поскольку для осуществления протективного эффекта ИЛ-1ra должно быть заблокировано более 95 % рецепторов к ИЛ-1 [4, 5].
Баланс ИЛ-1 и ИЛ-1ra лежит в основе регуляции воспалительного ответа. Величина продукции ИЛ-1ra тесно связана с генетическими факторами; продемонстрировано значение аллеля-2 ИЛ-1ra (IL1RN-2) как предиктора развития заболеваний почек [6]. Так, носители аллеля IL1RN-2 отличаются низкой продукцией (низкие продуценты) защитного ИЛ-1ra моноцитами в ответ на воспалительные стимулы, но при сохранении уровня продукции провоспалительного ИЛ-1 [7]. У гомозиготных носителей этого аллеля отмечается прогрессирующее течение гломерулонефрита и диабетической нефропатии с быстрым формированием терминальной почечной недостаточности – в среднем через 1,5 и 2,2 года соответственно [8, 9]. В исследовании V. Rauta у больных IgA-нефропатией уровень экскретируемого с мочой ИЛ-1ra был ниже, чем у здоровых лиц. Уровень экскреции ИЛ-1ra и ИЛ-1 не коррелировал с величиной протеинурии, длительностью болезни и гистологической картиной в биоптатах почки. Однако при более высоком индексе ИЛ-1ra/ИЛ-1 процессы накопления мезангиального матрикса, а также степень интерстициального воспаления и фиброза были менее выражены, чем при низком
или нормальном соотношении ИЛ-1ra/ИЛ-1β [10]. Кроме того, высокая концентрация ИЛ-1ra в сыворотке крови и моче больных протеинурическими формами IgA-нефропатии определяли хороший ответ на иммуносупрессивную терапию и меньший риск нарушения функции почек [11]. В исследовании G. Sturfelt и соавт. выявлены также низкие значения ИЛ-1ra в сыворотке крови больных с волчаночным нефритом в отличие от повышенных концентраций ИЛ-1ra у больных СКВ без поражения почек [12].
При введении ИЛ-1ra экспериментальным животным с тяжелым прогрессирующим анти-БМК-нефритом уменьшалось количество полулуний, капиллярных тромбов в клубочках, степень гломерулосклероза, а также тубулярной атрофии и интерстициального фиброза, что сопровождалось снижением протеинурии и сохранением функции почек [13]. Протективное противовоспалительное и антифиброгенное действие ИЛ-1ra наиболее выражено в тубулоинтерстициальном отделе почки, где отмечаются торможение экспрессии адгезивной молекулы ICAM-1 и уменьшение макрофагальной инфильтрации в эндотелии перитубулярных капилляров и на поверхности тубулярных клеток [14].
Тканевые ингибиторы матриксных металлопротеиназ (ТИМП)
Как показали исследования последних лет, ТИМП являются многофункциональными протеинами и способны оказывать разнообразные эффекты, связанные главным образом с регуляцией активности основных протеолитических ферментов – матриксных металлопротеиназ (ММП) [15]. ММП играют
ключевую роль в расщеплении компонентов экстрацеллюлярного матрикса (ЭЦМ), базальных мембран и цитоскелета клеток. Субстратом действия ММП помимо матриксных белков являются цитокины, факторы роста и их рецепторы, молекулы клеточной адгезии, хемокины, что объясняет регулирующую функцию системы ММП/ТИМП в механизмах воспаления, фиброза, в межклеточных и клеточно-матриксных взаимодействиях, клеточной миграции, в процессах эпителиально-мезенхимальной трансдифференциации [16, 17]. Установлены эффекты ТИМП, напрямую не связанные с ингибированием протеолитической активности ММП, в частности участие в регуляции процессов клеточного роста, пролиферации, апоптоза.
В физиологических условиях в почке функционирует сбалансированная система ММП/ТИМП. Нарушение соотношения компонентов этой системы может быть одним из патогенетических механизмов развития ряда острых и хронических заболеваний почек, в т. ч. и ХГН [18]. На определенных этапах увеличение
экспрессии в ткани почки ТИМП можно рассматривать как адаптивное, направленное на ограничение провоспалительных эффектов ММП. Увеличение уровня ТИМП, сопровождающееся снижением ММП, ассоциируется с усиленным накоплением фиброза в клубочках и особенно в интерстиции почки, что
приводит к развитию почечной недостаточности. В последние годы появилась возможность определения отдельных факторов системы протеолиза в моче больных ХГН, но исследования такого направления пока единичны.
Нами был проведен комплексный анализ изменений уровня в моче ММП и их ингибиторов (ТИМП и ПАИ-1) на разных стадиях течения ХГН в сопоставлении с выраженностью морфологических изменений в почке [19]. Мы установили, что с нарастанием активности заболевания (высокая протеинурия, нефротический синдром, остронефротический синдром) отмечается однонаправленный характер изменений всех факторов – увеличение в моче ММП и их ингибиторов (ТИМП и ПАИ-1), отличающееся только количественно. По нашему мнению, такие однонаправленные изменения носят адаптационный характер в условиях активного воспаления в почке. У больных ХГН со стойкой почечной недостаточностью (ПН) был отмечен дисбаланс в системе протеолиза, характеризующийся резким снижением уровня в моче ММП, ТИМП и непропорционально высокой активностью ПАИ-I. Такой
спектр мочевых биомаркеров, по нашему мнению, отражает дезадаптационные изменения в почке – ослабление механизмов протеолиза, усиление фиброгенеза, приводящее к развитию почечной недостаточности.
Полученное в последние годы подтверждение важной роли компонентов системы ММП/ТИМП в механизмах воспаления и фиброза в почке обосновывает новые подходы к нефропротекции путем целенаправленного воздействия на ММП и ТИМП, в частности, через нивелирование эффектов основных провоспалительных и профиброгенных цитокинов – ангиотензина II и трансформирующего фактора роста β1.
Инактиваторы лейкоцитов и резидентных клеток
В период выздоровления при остром гломерулонефрите инфильтрирующие почку воспалительные лейкоциты подвергаются инактивации, взаимодействуют с медиаторами защиты – противовоспалительными цитокинами и биоактивными липидами, играют важную роль в предотвращении персистирования (хронизации) воспаления.
Интерлейкин-10
Интерлейкин-10 (ИЛ-10) (молекулярный вес – 37кДА) многофункциональный противовоспалительный цитокин. Главными источниками ИЛ-10 in vivo являются лимфоциты и макрофаги [20], а также активированные резидентные клетки, аутокринным путем подавляющие продукцию различных воспалительных цитокинов соседними клетками [21]. Секреция ИЛ-10 начинается через несколько часов после действия провоспалительных факторов – иммунных комплексов, липополисахаридов, простагландинов, катехоламинов, цитокинов, главным образом – ФНО-α [22, 23].
Основные иммунологические эффекты ИЛ-10 связаны с регуляцией баланса Т-хелперов первого типа/Т-хелперов второго типа (Th1/Th2) [20]. Активация Th1 через секрецию ФНО-α, ИЛ-2 и интерферона-γ ведет к стимуляции главным образом Т-лимфоцитов и макрофагов и развитию клеточного типа ответа (Т-клеточная цитотоксичность, активация макрофагов, клеточноопосредованное воспаление), тогда как Th2-цитокины (ИЛ-4, ИЛ-5, ИЛ-6, ИЛ-10, ИЛ-13 и ИЛ-25) имеют противовоспалительные свойства,
ингибируя активацию макрофагов, пролиферацию Т-клеток и продукцию провоспалительных цитокинов, стимулируя гуморальное звено иммунитета – синтез антител IgE, IgA, некомплементарное связывание IgG [24]. Продукцию ИЛ-10 помимо Th2 осуществляют особые регуляторные противовоспалительные Т-клетки (CD4+, CD25+ и Tr1). Повышение уровня ИЛ-10 подавляет Th1-клеточно-опосредованное повреждение и сдвигает баланс Th1/Th2-преимущественно в сторону Th2-противовоспалительных цитокинов [25].
Помимо влияния на баланс Т-хелперов ИЛ-10 ингибирует в мононуклеарных клетках продукцию воспалительных цитокинов, хемокинов, адгезивных молекул, реактивных радикалов кислорода и простагландинов, литических ферментов моноцитов – матриксных металлопротеиназ, супероксид-
анионов [26, 27, 28].
Противовоспалительный эффект ИЛ-10 был оценен в экспериментальных моделях гломерулонефрита. У крыс с нефротоксическим сывороточным нефритом, профилактически получавших инъекции ИЛ-10 за сутки до инициации нефрита, величина протеинурии была на 75 % ниже, чем в контроле [29]. В модели с анти-БМК-нефритом у мышей назначение ИЛ-10 оказывало не только профилактический, но и лечебный эффект. Рекомбинантный ИЛ-10 предотвращал образование полулуний, отложение фибрина в клубочках, накопление Т-лимфоцитов и макрофагов в ткани почки, что приводило к
снижению протеинурии и креатинина сыворотки [30].
Результаты применения ИЛ-10 при экспериментальном анти-Thy1-мезангиопролиферативном нефрите подтвердили не только противовоспалительную (уменьшение экспрессии ИЛ-1β и ICAM-1 в клубочках), но и антипролиферативную роль ИЛ-10 (уменьшение пролиферации мезангиальных клеток) [31].
В экспериментальных моделях нефротоксического нефрита и анти-БМК-нефрита активация Th1-клеток и уменьшение продукции ИЛ-10 ведут к клеточно-опосредованному повреждению клубочка – часто с формированием полулуний (экстракапиллярная пролиферация, накопление моноцитов и Т-клеток) [32]. У человека тяжелые и быстропрогрессирующие формы ГН с морфологической картиной анти-БМК-нефрита с полулуниями, ANCA-ассоциированого ГН, редкие случаи мембранозной нефропатии с формированием полулуний характеризуются активацией преимущественно Th1-зависимого механизма гиперчувствительности замедленного типа с дефицитом ИЛ-10 [33, 34, 35]. При пролиферативных формах нефрита, в т. ч. при диффузном пролиферативном волчаночном нефрите, выявляется более низкий, чем при непролиферативных формах (мембранозная нефропатия, нефрит с минимальными изменениями), уровень цитокинов Th2, включая ИЛ-10 [36]. Таким образом, у больных с преимущественным Th1-клеточным иммунным ответом и дефицитом противовоспалительных,
антипролиферативных цитокинов, в первую очередь ИЛ-10, развиваются тяжелые прогрессирующие формы поражения почек – часто с формированием полулуний, требующих наиболее активного лечения.
С учетом эффективности применения ИЛ-10 при экспериментальном нефрите можно предполагать положительные результаты и в лечении гломерулонефрита у человека. Особенно это касается нефритов с тяжелым прогрессирующим течением с доминированием Th1-клеточного иммунного ответа
(морфологически – пролиферативные формы с образованием полулуний).
Протективная роль регуляторных иммунных клеток при повреждении почечной ткани
Исследованиями последних лет продемонстрирована не только повреждающая, но и протективная роль клеток почечного инфильтрата (Т-лимфоцитов, макрофагов и дендритных клеток) в торможении гломерулярного и тубулоинтерстициального воспаления. В некоторых экспериментальных моделях заболеваний почек установлено, что отдельные подтипы CD4+ Т-хелперных клеток являются протективными или регуляторными: мигрируя в очаг повреждения, они блокируют патогенные подтипы Т-клеток, уменьшают воспаление и тормозят развитие заболевания [37]. Позже была показана принадлежность этих клеток к субпопуляции CD4+CD25+ (Tрег), участвующей в механизме формирования иммунологической толерантности к собственным антигенам и обеспечивающей негативный контроль иммунного ответа, в т. ч. аутоиммунного. При заболеваниях почек “адаптивные”
Трег могут оказывать супрессивное действие на эффекторные и цитотоксические Т-лимфоциты почечного инфильтрата при непосредственном клеточном контакте, а также путем продукции противовоспалительных цитокинов и усиления апоптоза [38, 39]. Более того, Трег снижают активацию и пролиферацию других клеток воспалительного инфилтрата – макрофагов дендритных клеток, В-, NK-клеток и нейтрофилов [40].
Хорошо известна роль в механизмах прогрессирования заболеваний почек макрофагов, однако появились данные о том, что макрофаги могут иметь и протективное значение, участвуя в процессе репарации почечной ткани после повреждения. Эти макрофаги относятся к “альтернативному”
фенотипу – М2. Активация макрофагов по альтернативному пути происходит под действием Th2-цитокинов – ИЛ-4 или ИЛ-13 (М2а), а также иммунных комплексов в комбинации с ИЛ-1β или липополисахаридами (М2в) или при добавлении ИЛ-10, ТФР-β, глюкокортикоидов (М2с)[ 41]. В результате формируется М2-иммуногрегуляторный/иммуносупрессивный фенотип, участвующий в разрешении воспалительного процесса и репарации [42]. Наряду с подавлением секреции
провоспалительных медиаторов иммунными клетками М2 характеризуются повышенной способностью к фагоцитозу, усиленной продукцией противовоспалительных и трофических факторов: ИЛ-1ра, аргиназы-1 (тормозящей продукцию NO), ТФР-β [43, 44].
Противовоспалительные эйкозаноиды
Липоксины
В последние годы появились данные, свидетельствующие об участии в резолюции воспаления эндогенных липидных медиаторов с противовоспалительными свойствами. В то время как в начальной фазе иммунного ответа преобладают липидные медиаторы воспаления (лейкотриены, простагландины), в дальнейшем происходит “переключение” на противовоспалительные липоксины. Липоксины – продукты метаболизма арахидоновой кислоты, которые продуцируются локально в очаге воспаления в процессе различных межклеточных взаимодействий: при контакте нейтрофилов с эпителиальными клетками или тромбоцитами происходит активация липоксигеназы (ЛО) с образованием липоксинов А4 и В4 [45]. При приеме аспирина ацетилирование циклооксигеназы-2 на
эндотелии в очаге воспаления приводит к синтезу аспирининдуцированных липоксинов (АИЛ) с более высоким, чем нативные липоксины, потенциалом – 15-эпи-липоксин А4 или 15-эпи-липоксин В4 [46].
Потенциальными активаторами синтеза липоксинов при межклеточных взаимодействиях являются Th2-цитокины – ИЛ-4 и ИЛ-13, повышающие экспрессию ЛО на моноцитах и эпителиальных клетках [47, 48]. Полагают, что липоксины выполняют роль эндогенных “стоп”-сигналов для миграции нейтрофилов в очаг воспаления [49]. Нативные липоксины, АИЛ и их синтетические аналоги ингибируют все этапы миграции нейтрофилов в очаг воспаления – хемотаксис, адгезию к эндотелиальным клеткам, “качение” вдоль слоя эндотелия, трансмиграцию через эндотелиальные клетки [50]. По-видимому, противовоспалительный потенциал липоксинов очень широк, т. к. назначение синтетического аналога 15-эпи-липоксина А4 в ранней фазе экспериментального анти-БМК-нефрита вело к ингибиции транскрипции 21-го гена провоспалительных медиаторов [51]. Липоксины способствуют формированию макрофагов М2, приводя к повышению их фагоцитарной активности, не сопряженной с высвобождением провоспалительных цитокинов [52], обладают антипролиферативным действием на мезангиальные клетки клубочков и являются потенциальными ингибиторами сосудистого эндотелиального фактора роста (VEGF) [53, 54]. Кроме того, липоксины могут повышать почечный кровоток и уровень клубочковой фильтрации [55].
Продукция липоксинов была изучена при остром постстрептококковом гломерулонефрите (ОГН) у детей – ярком примере спонтанного выздоровления, при котором эндогенные механизмы самозащиты оказываются состоятельными, характеризуясь адекватным синтезом противовоспалительных факторов. Начало разрешения воспаления приходилось на пик экспрессии 15-ЛО и липоксина А4 (LXA4), по времени совпадало со значительным снижением количества лейкотриена В4 (LTB4) и инфильтрации ткани почки нейтрофилами [56].
В отличие от ОГН при хроническом течении гломерулонефрита отмечается уменьшение индекса LXA4/LTB4, свидетельствующее о выраженном дисбалансе между этими факторами [57]. Так, у больных с развитием нефрита при пурпуре Шенлейн –Геноха экспрессия 15-ЛО в ткани почки, а также уровень LXA4 в сыворотке крови и моче были ниже, чем у больных без поражения почек, эти показатели коррелировали с тяжестью нефрита [58].
Белки теплового шока
Белки теплового шока (БТШ) – внутриклеточные высокостабильные белки, которые контролируют образование и обмен других внутриклеточных белков и участвуют в поддержании целостности клетки, функционируя как молекулярные шапероны. БТШ объединяются в семейства по молекулярному весу:
малые БТШ (16–40кДа), БТШ 60кДа, БТШ 70кДа, БТШ 90кДа, 110БТШкДа. Наряду с конституциональными БТШ, которые экспрессируются в норме в количестве 5–10 % всех белков
клетки, существуют индуцибельные БТШ, составляющие до 15 % всех клеточных белков, синтез которых повышается в ответ на различные виды повреждающего внешнего воздействия: температурный, оксидантный, токсический, осмотический, а также воспалительный стресс. В ткани почки в норме экспрессируются многие БТШ, уровень их экспрессии изменяется после повреждения [59]. Так, экспрессия БТШ 70 резко возрастает при ишемической и токсической почечной недостаточности [60, 61], а также после ишемии-реперфузии [62]. Повышение уровня БТШ внутри клетки при повреждении играет роль в восстановлении структуры частично денатурированных протеинов и в деградации белков, не подлежащих восстановлению [63].
Цитопротективная функция низкомолекулярного БТШ 27 обусловлена взаимодействием с актиновым цитоскелетом, что обеспечивает стабилизацию актиновых волокон клетки в условиях воздействия (стресса), например, под влиянием ФНО-α [64]. Структура ножек подоцитов – неотъемлемой части фильтрационного барьера почки – напрямую зависит от состояния актиновых микрофиламентов и процессов их полимеризации, регулируется БТШ 27. Недостаточная экспрессия БТШ 27 в подоцитах может приводить к утрате нормальной структуры фильтрационного барьера клубочков почек и развитию протеинурии [65].
Таким образом, увеличение синтеза БТШ внутри клетки в ответ на различные повреждающие факторы, в т. ч. воспалительные, является адаптивным защитным механизмом, повышающим резистентность клеток к клеточному стрессу и предотвращающим гибель клеток за счет стабилизации и восстановления поврежденных белковых молекул [66]. БТШ могут высвобождаться во внеклеточную среду или экспрессироваться на поверхности клеток и таким образом контролировать воспаление [67].
Роль БТШ в Т-клеточной регуляции хронического воспаления
Белки теплового шока, особенно белки семейства 60 и 70, относятся к иммунодоминантным молекулам, т. е. пептидные последовательности микробных БТШ 60 и 70 являются основными эпитопами, стимулирующими противоинфекционный иммунный ответ. Для БТШ прокариотических (бактерий) и
эукариотических клеток (млекопитающих и человека) характерна высокая степень гомологии, достигающая 50–60 % (как, например, для семейства БТШ 60) [68]. Это может означать, что белки теплового шока являются потенциальными кандидатами для молекулярной мимикрии и способны
играть роль в развитии не только противоинфекционного иммунитета, но и аутоиммунитета [69]. Данные о повышении уровня БТШ 60 и 70 при иммунных, в т. ч. аутоиммунных заболеваниях – диабете, ревматоидном артрите, склеродермии, а также при трансплантации органов, свидетельствуют
в пользу этого предположения [70, 71]. В исследовании Н.А. Мухина и соавт. уровень антител к БТШ 70 были значительно выше у больных с системной красной волчанкой, дерматомиозитом и хроническим гломерулонефритом, чем у здоровых лиц [72].
Первым доказательством того, что БТШ оказывают противовоспалительный эффект, т. е. играют протективную роль при иммуновоспалительных заболеваниях, были результаты исследования с введением экспериментальным животным с аутоиммунным адьювантным артритом БТШ 60, свидетельствующие о последующем торможении развития заболевания [73].
При воспалении и повреждении клеток внутриклеточно расположенные белки теплового шока могут экспрессироваться на их поверхности или выделяться во внеклеточное пространство. Эпитопы БТШ распознаются Т-клетками, что приводит к формированию регуляторных противовоспалительных фенотипов реактивных Т-клеток [74]. В результате происходит “переключение” Th1-фенотипа на Th2 c повышением уровня противовоспалительных ИЛ-10 и ИЛ-4 в Т-лимфоцитах [75].
Противовоспалительный потенциал БТШ при заболеваниях почек мало изучен. Выявлено повышение внеклеточной экспрессии БТШ при экспериментальном нефрите [76], а также у больных с различными формами нефрита: при нефрите с минимальными изменениями, ФСГС; мембранозной нефропатии, пролиферативных формах гломерулонефрита, включая волчаночный нефрит [63, 77].
Заключение
Исследования последних лет были сосредоточены в основном на повреждающих механизмах развития заболеваний почек, однако в ткани почки существует многоуровневая конституциональная защитная система, экспрессирующая разнообразные медиаторы защиты, с помощью которых эта система противостоит факторам повреждения. Протективные механизмы, т. н. индуцированная защита, являются неотъемлемым компонентом процесса воспаления. Благодаря активации этой системы влияние воспалительных цитокинов (ИЛ-1, ФНО-α и др.) ограничивается соответствующими ингибиторами или
взаимодействующими цитокинами. Для системы самозащиты в почке характерно формирование каскадов защитных молекул. Например, ИЛ-10 может повышать экспрессию нескольких протективных ИЛ-1ра и рецептора к ФНО-α, а также БТШ в ткани почки. ИЛ-4 и ИЛ-13 стимулируют продукцию ИЛ-1ра и липоксинов. Главным источником противовоспалительных факторов, ограничивающих дальнейшее распространение воспаления, являются “альтернативные” клетки воспалительного
инфильтрата почки: регуляторные Т-лимфоциты (CD4+CD25+) и “альтернативные” М2-макрофаги. В основе противовоспалительного действия экспрессирующихся на поверхности БТШ (60 и 70) лежит активация регуляторных клеток инфильтрата и секреция ими противовоспалительных цитокинов.
Своевременная реализация противовоспалительной составляющей процесса воспаления – залог успешного его разрешения и выздоровления. Например, значительное повышение количества липоксинов в почке в период выздоровления при ОГН в эксперименте и у человека позволяет отнести их к важным молекулам-регуляторам, способным ограничить воспалительный процесс. Несостоятельность (недостаточность) эндогенных механизмов самозащиты почки приводит к персистированию эффекторного, повреждающего звена воспаления и способствует прогрессирующему течению болезни. О дисбалансе между про- и противовоспалительными
факторами при хронических прогрессирующих заболеваниях почек можно судить по индексам ИЛ1ра/ИЛ-1β, ММП/ТИМП, а также LXА4/LTВ4 в моче.
Усиление эндогенных защитных механизмов путем введения ключевых протективных факторов, способных модулировать воспаление в почке и “переключать” этот процесс в сторону ограничения/резолюции, представляет собой перспективное направление терапевтического воздействия при прогрессирующих заболеваниях почек.


