
Термин “цилиопатии” – новый, существует только с начала ХХI в. По мнению J.L. Badano et al (2006) [1], “цилиопатии – это появляющийся класс заболеваний человека, связанных с генетическими нарушениями”. К цилиопатиям относят множество различных болезней, как правило, клинически
имеющих проявления кистоза почек.
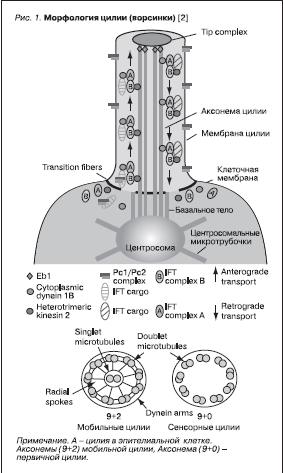
Ворсинки (цилии) и жгутики известны с ранних этапов эволюции живых организмов, они служили одноклеточным средством передвижения. По мере адаптации растений к земному обитанию у них произошла практически полная потеря этих органелл. Однако сложноорганизованные живые существа, включая всех млекопитающих, использовали цилии для выполнения разнообразных функций. Оказалось, что белки, входящие в состав ворсинок, – весьма консервативны и сохраняли свои свойства на протяжении многих миллионов лет [2]. В настоящее время определено, что ворсинка представляет собой высокоорганизованную органеллу. Структурной единицей ворсинки является аксонема, состоящая из 9 пар микротрубочек. Существует два вида цилий: мобильные (Motile cilium) и немобильные, сенсорные (Sensory cilium) (рис. 1, 2). Для сенсорных цилий характерно еще название “первичные”, или “моноцилии”. Первичные цилии у млекопитающих вездесущи: они обнаружены на обонятельных клетках, палочках и колбочках сетчатки, клетках почечного тубулярного эпителия, мезенхимальных клетках, нейронах. В состав замыкательной части аксонемы входят полицистины – продукты генов поликистозной болезни почек, продукты генов нефронофтиза (НФФ), рецепторы соматостатина, серотонина, ангиопоэтина, тромбоцитарного фактора роста-α, ванилоида-4, представляющего рецепторные каналы с переменным потенциалом [3, 4]. Длина первичных ворсинок (цилия) связана с количеством тубулина – продукта гена туберозного склероза [5].
Рисунок 1. Морфология цилии (ворсинки) [2]
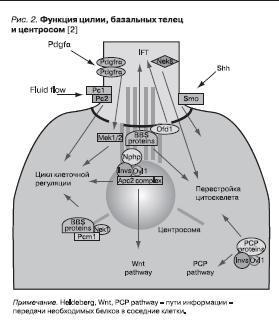
Рисунок 2. Функция цилии, базальных телец и центросом [2]
С 1930-х гг. пристально изучается синдром Картагенера, зависящий от нарушений мобильных цилий [6]. Заболевание носит также название “синдром неподвижных ресничек”, т. к. цилии в переводе звучат как ворсинки и реснички, а синдром Картагенера характеризуется наличием трех составляющих: бронхоэктазов, хронического синусита а также обратного расположения внутренних органов в 50 % случаев. В основе патологии лежит генетически детерминированная дискине-
зия мобильных ворсинок, диагностикой и лечением которой занимаются пульмонологи и оториноларингологи.
В конце ХХ – начале ХХI вв. с нарастающей частотой публикуются работы, говорящие о цилиях как причине развития разнообразных заболеваний, причем акцент делается на состоянии т. н. первичных цилий, или немобильных ворсинок [7]. Генетические исследования показывают, что многие кистозные заболевания почек и печени связаны с мутацией генов, определяющих состояние первичных ворсинок в почках [8]. Сообщается об участии ворсинок в развитии заболеваний, когда страдание касается одного органа, в частности почек, и о плейотропных болезнях, таких как синдром Барда–Бидля [9], при котором практически все органы в той или иной степени поражены в связи с изменением структуры или функции цилий [10].
Первичные цилии встречаются во всех органах, судя по исследованиям последних лет. По мере изучения структуры и функции ворсинок оказалось, что движение подвижных ворсинок осуществляется “вперед–назад”. Неподвижные (immotile cilia), первичные ворсинки не являются полностью неподвижными, но их движение ограничивается ротацией вокруг своей оси. Ворсинка содержит сотни различных белков, и изменение структуры или функции одного из них сопровождается выходом из строя всей цилии [11].
Cilia обладают активным обменом веществ. Каждые 6 часов происходит обмен около 20 % белков ворсинки с участием моторных белков кинезинов. Этот обмен белков получил название внутрижгутикового транспорта [12]. Суть указанного процесса – в переносе структурных белков ворсинки из цитоплазмы в микротрубочки, где эти белки включаются в процесс структурной сборки аксонемы [13]. Нормального процесса структурного обновления аксонемы не происходит, если дефектны киназы, о чем говорят экспериментальные исследования [14].
Одним из важнейших свойств цилий является их влияние на формирование лево-правой асимметрии тела. В настоящее время расшифрован механизм влияния цилий на процесс развития асимметрии тела у позвоночных. Эмбриональный узел, расположенный в дистальной части зародыша, является
организующей структурой на ранних этапах его развития. При этом эмбриональный узел имеет единственную первичную цилию, осуществляющую передачу импульса на периферию [15]. Происходит первоначальное нарушение биполярной симметрии эмбриона, вслед за чем устанавливается асимметрия экспрессии генов эмбрионального узла. Это приводит к передаче импульса к наружному листку мезодермы и развивающимся органам. Предполагают, что в основе этого процесса лежит ток жидкости, окружающий эмбриональный узел. Выдвигаются гипотезы относительно механизма влияния “узлового потока”. Согласно наиболее распространенной гипотезе, возникло представление, будто мобильные ворсинки, находящиеся в центре узла, генерируют ток жидкости, а первичная ворсинка, расположенная на периферии эмбрионального узла, улавливает этот ток и влияет на последующий каскад явлений, обусловливающих асимметрию органов. Возможно возникновение ситуации, когда нарушено движение узлового потока, что может произойти при неправильном движении ворсинки, тогда формирование лево-правой асимметрии приобретает “случайный характер”. Часть эмбрионов в эксперименте имеют situs solitus, т. е. нормальное расположение внутренних органов. Другая часть экспериментальных эмбрионов получает situs inversus – обратное расположение органов. Подобный результат измененных в морфологическом или функциональном состоянии ворсинок на формирование лево-правой асимметрии органов, изученный в эксперименте на крысах [16], оказался типичным для всех позвоночных. У человека подобная аномалия отмечается при инфантильной форме НФФ.
В ХХI в. стало очевидно, что первичные цилии вездесущи в теле млекопитающих и действуют как клеточные механо-сенсорные органеллы [17]. Количество цилий отличается в клетках различных органов. В некоторых эпителиальных клетках может выявляться несколько сотен ворсинок с аксонемой
9 + 2, в то же время в той же клетке имеется лишь одна цилия с аксонемой 9 + 0. Хотя в настоящее время стало ясно, что все цилии являются своеобразными сенсорами для клетки, однако именно первичные цилии рассматриваются как антенны, воспринимающие внешние химические и механические воздействия [18]. Именно их называют “сенсорными цилиями”. Они улавливают звук, свет, запах, движение жидкости, изменения осмомолярности. Полученная информация передается
своеобразным комплексом внутриклеточных органелл: цилии/базальные тельца/центросома внутри и вне клетки с помощью ионов кальция. Исследования последних лет указывают на незаменимую роль цилий в физиологии и гомеостазе эпителиальных почечных клеток [19].
К первичным цилиям (ворсинкам) привлечено внимание в различных аспектах теоретической и клинической медицины [20]. Обращено внимание на их роль в развитии эукариот и участие в формировании болезней. В осуществлении сложных функций участвует не только первичная ворсинка, но и комплекс ворсинка–базальные тельца и центросома [21].
Структура цилии очень сложна. Каждая аксонема имеет свою оболочку, которая отделяет ворсинку от содержимого самой клетки. Характер белковых структур, передающих полученную информацию внутри клетки (обозначаемое как signaling) и в соседние клетки (называемые pathway), постепенно расшифровывается. Большая часть этих бел-
ковых структур содержит протеины, управление которыми осуществляется генами поликистозной болезни и НФФ. Появились работы, указывающие на роль цилий в формировании кист [22]. Гены НФФ оказываются в центре внимания генетиков [23].
НФФ – рецессивно передающееся наследственное заболевание, которое среди генетически детерминированной патологии наиболее часто приводит к хронической почечной недостаточности (ХПН) в детском или раннем взрослом состоянии [24]. Позиционное клонирование девяти генов (NPHP 1–9), ответственных за формирование НФФ, и изучение функции белков (нефроцистинов), кодирование которых осуществляется указанными девятью генами, привели к заключению, что кистозные болезни почек могут быть названными “цилиопатиями” [25]. Хотя кистозные болезни у человека гетерогенны, исследования генетики и молекулярной биологии показывают, что в их развитии немаловажную роль имеют продукты генов моноцилий [26]. Показано, что продукты генов НФФ экспрессируются на первичных цилиях, чувствительных органеллах клеток, на базальных
тельцах, связанных с цилиями, и центросомах, а т. к. они присутствуют во всех органах и тканях, этим объясняется частое сочетание поражения почек при НФФ со страданием других органов. Наиболее изучен Senior–Loken syndrome, для которого характерна тареторетинальная дегенерация органа
зрения, проявляющаяся врожденной слепотой. В последнее время стало известно, что мутирующий ген при синдроме Senior–Loken – NPHP-5 [27].
НФФ имеет место при синдроме Когана, для которого характерна окулярная, моторная апраксия при синдроме Жуберта, при котором наблюдается колобома/дегенерация сетчатки и аплазия мозжечка. При синдроме Майнца–Салдино кроме НФФ имеются фиброз печени и мозжечковая атаксия.
НФФ описан в разных странах мира [28]. Его частота на различных территориях не одинакова : 9 больных на 1 млн популяции в США, 1 – на 50 тыс. живорожденных в Канаде. При этом в США 5 % детей с терминальной ХПН связаны именно с НФФ [29]. Морфологически для НФФ характерны наличие кортикомедуллярных цист, изменения базальных мембран и выраженные проявления тубулоинтерстициального процесса (рис. 3, 4).
Рисунок 3. УЗИ почек Алины Б. 13 лет. Диагноз: Нефронофтиз ювенильный
Рисунок 4. Морфология почки при нефронофтизе: выражены кисты, интерсциальные изменения, фиброз
Клинически обычно выделяют три типа заболевания НФФ:
• ювенильный,
• инфантильный,
• взрослый.
Нефронофтиз 1-го типа – ювенильный, при котором мутантный ген NPHP I локализован на длинном плече хромосомы 2 (2q12-q13), продуктом гена является нефроцистин-1. Заболевание приводит к ХПН в среднем в 13 лет (от 7 до 25). Заболевание развивается постепенно с появления полиурии,
полидипсии, анемии.
Нефронофтиз 2-го типа – инфантильный, мутантный ген INV локализован на длинном плече хромосомы 9 (9q22-q31). Продуктом гена является инверсин. Особенностью 2-го типа НФФ является увеличение размеров почек и появление кист в медуллярной и кортикальной зонах. Так как в функции
инверсина входит правильное расположение внутренних органов в период раннего эмбриогенеза, следствием мутации гена является не только развитие НФФ, но и появление situs viscerum inversus.
Нефронофтиз 3-го типа – ювенильный, для него характер- на мутация гена NPHP 3, картированного на длинном плече хромосомы 3 (3q21-q22). Продукт гена – нефроцистин-3. ХПН развивается в среднем в 19 лет (от 11 до 28). Описан в большой венесуэльской семье. Нефронофтиз 4-го типа (взрослый), при котором происходит мутация гена NPHP 4, расположенного на коротком плече хромосомы 1 (1р36), приводит к развитию ХПН в среднем в 22 года (от 11 до 34 лет). Продукт гена – нефроцистин-4.
Проведено позиционное клонирование более чем у 1000 семей с НФФ и установлено, что наиболее частой причиной НФФ оказывается мутация NPHP1 – гена, ответственного за 1-й тип НФФ (21 %). Из остальных 8 генов наиболее часто встречается NPHP5 (3 %), оставшиеся выявляются еще реже.
В общей сложности более чем в 70 % случаев НФФ установить мутацию одного из девяти изученных генов NPHP не удается (рис. 5).
Рисунок 5. Частота мутации генов NPHP среди 1079 больных нефронофтизом в различных странах мира, % [23]
Возникает вопрос о наличии еще неидентифицированных генов, ответственных за развитие НФФ [23].
Это тем более очевидно, что идентификация NEK8-мутации как проявление NPHP 9 произошла только
в 2008 г. [30].
Основываясь на изменениях нефроцистина 3 в эксперименте на мышах, которым свойственно развитие НФФ, осуществлено исследование семей, у которых отмечена мутация NPHP 3. Причем определение NEK8 первоначально проводилось в эксперименте на мышах, а затем было проверено наличие NEK8-мутации в 588 случаях НФФ с контролем в 80 семьях, где не было случаев НФФ. Исследования
осуществлялись в Швейцарии, Австрии и Курдистане. В Курдистане заболевание зафиксировано в случае кровнородственного брака. Генетические и морфологические исследования проведены у всех членов семей. Оказалось, что потеря функциональной активности нефроцистина 3 приводит к различным патологическим состояниям. Среди них были летальность в эмбриональном периоде, синдром Meckel–Gruber, обратное расположение внутренних органов и почечно-печеночно-панкреатическая дисплазия (рис. 6). Наиболее типичным оказалось наличие CACUT-синдрома, т. е. сочетание аномалии почек и органов мочевыведения [31].
Рисунок 6. Сочетанное поражение нескольких органов при наличии в семье мутаций гена NPHP-3 [31]
Параллельно с изучением роли ворсинок в развитии НФФ осуществляется определение их влияния на формирование поликистозной болезни почек [32]. Современные исследования показывают, что существует прямая связь между механосенсорной функцией белков первичных ворсинок и возможностью развития поликистозной болезни почек (ПКБ) [33]. ПКБ в аутосомно-доминантном (АД) и аутосомно-рецессивном (АР) вариантах является одним из наиболее распространенных генетически детерминированных заболеваний человека, которое ведет к развитию почечной недостаточности. АДПКБ нередко называют “ взрослым типом” болезни [34]. Это связано с тем, что основные клинические проявления болезни становятся явными по мере взросления человека и ХПН развивается не
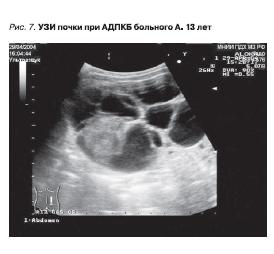
ранее 30–40 лет. Однако использование УЗИ плода показывает, что в эмбриональном периоде уже можно заподозрить наличие ПКБ, а первые клинические проявления видны у ребенка (рис. 7).
Рисунок 7. УЗИ почки при АДПКБ больного А. 13 лет
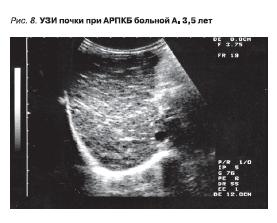
АРПКБ чаще проявляется уже в первые месяцы жизни ребенка, причем одним из обычных осложнений бывают нарушения со стороны дыхательного тракта. Однако возможно и более позднее обнаружение
АРПКБ. Яркую картину представляет УЗИ почек (рис. 8).
Рисунок 8. УЗИ почки при АДПКБ больной А. 3,5 лет
Нарушения функционального или структурного состояния цилий канальцевого эпителия в эмбриональном периоде развития ребенка приводят к нарушению правильной ориентации тубулярных клеток. Развивается синдром, носящий название “defective planar cell polarity”, который является одним из основных в формировании ПКБ [35]. В развитии ПКБ велика роль продуктов генов поликистозной болезни полицистина 1, полицистина 2 и фиброцистина [36]. При изучении модели животных с близкой по структуре почки человека (orthologous animal model) в условиях острого поражения
происходит нарушение передачи информации (необходимых белков как внутри клетки, так и между клетками), что чревато развитием поликистозной болезни почек [37]. Дефекты межклеточного взаимодействия при поликистозной болезни проявляются заменой E-катгерина на фетальный N-катгерин,что ухудшает возможности реагирования β-катенина с другими связывающими актин белками [38]. В норме полицистин, белки, кодируемые генами НФФ, и β-катенин представляют барьер для ненаправленного тока жидкости и ионов. Усиленный ток жидкости и ионов происходит в случаях ПКБ и приводит к дилатации канальцев. Об этом можно судить по экспериментальным данным, связанным с нарушениями структуры или функции первичных цилий в эмбриональном периоде.
Немаловажную роль в указанном процессе играют гены, кодирующие белки, входящие в состав аксонемы цилии [39]. Кистозным изменениям канальцев сопутствует увеличение внеклеточного матрикса и развитие интерстициального фиброза, причем характер интерстициального фиброза при
ПКБ не отличается от того, что наблюдается при других заболеваниях почек. Естественно, что одним из методов терапевтического вмешательства оказываются ингибиторы АПФ (ИАПФ) и блокаторы ATII (бATII). Расчеты на снижение АД и стабилизацию кист в почках при использовании этих ренопротективных средств в терапевтических клиниках не оправдываются [40]. Однако лечение детей с АДПКБ эналаприлом (ИАПФ) в возрасте от 6 до 10 лет, чего не наблюдается в возрасте 17 лет (контрольная группа), приводит к стабилизации состояния кист, что вселяет определенный оптимизм [41].
В последние годы активно разрабатываются методы лечения ПКБ в эксперименте на животных. Имеется в виду воздействие на процессы, связанные со структурной или функциональной недостаточностью цист (рис. 9).
Рисунок 9. Схема влияния современных терапевтических средств при кистозах почек, обусловленных поражением цилий в эксперименте на животных [37]
Механизм действия этих препаратов показан в таблице.
Таблица. Современные методы лечения ПКБ в эксперименте на животных
Все случаи кистозов, а их на протяжении последнего десятилетия не менее 20, наблюдаемых в нефрологическом отделении МНИИ педиатрии и детской хирургии, хотя и относились к разным вариантам НФФ и ПКБ, имели некоторые общие черты. Это было раннее развитие болезни – от врожденного до выявленного в 6-летнем возрасте. У всех детей отмечалось наличие множества внешних стигм соединительнотканного дизэмбриогенеза и разнообразных аномалий развития внутренних органов (поражение глаз, пороки сердца, обратное расположение органов и др.). Характерным было первоначальное снижение тубулярных функций, а затем быстрое формирование
ХПН. Эти признаки в различных сочетаниях описываются при различных формах поликистозов [24].
Таким образом, не только в теоретической, но и в практической нефрологии постепенно формируется понимание, что кистозы связаны со структурным или функциональным нарушением первичных цилий (ворсинок), являющихся сенсорными органеллами практически всех органов млекопитающих. Их нарушения связаны с мутациями генов, кодирующих множество белков, ассоциированных с цилиями.
Изолированные кистозы приходится наблюдать при НФФ и поликистозных болезнях почек. В настоящее время еще невозможно клиническое выявление кистозов как цилиопатий, однако генетически это уже сделано. Задачей ближайшего будущего является не только осмысление новых терапевтических подходов к лечению кистозов, но и испытание имеющихся положительных результатов лечения экспериментальных кистозов у животных в клинической практике.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}