
Наличие нефритического синдрома (НиС) позволяет предположить наличие пролиферативных изменений в почечных клубочках, среди которых наиболее часто встречается мезангио- пролиферативный гломерулонефрит (ГН). Тем не менее само по себе понятие НиС не позволяет ни поставить правильный диагноз, ни назначить патогенетическое лечение, ни определить морфологическую основу заболевания. В связи с этим особое значение приобретает иммунофлюоресцентное исследование почечного биоптата. Чаще всего при этом удается выявить IgA- нефропатию (IgA-НП) - самое частое гломерулярное заболевание (ГЗ) во всех странах, где широко практикуется биопсия почки [1-2]. IgA-НП обнаруживается в 30-40 % всех биопсий почки в Азии, 15-20 % в Европе и 5-10 % в Северной Америке. Эти различия, вероятно, связаны с генетической предрасположенностью, географическими особенностями скрининговых исследований мочи и показаниями к биопсии. IgA-НП часто начинается в детском и подростковом возрасте. Некоторые авторы считают, что IgA-нефропатия и пурпура Шенлейн-Геноха - это клинические варианты одного заболевания [3]. Также при НиС могут диагностироваться мембранопролиферативный ГН (МПГН) и другие морфологические варианты.
Лечение НиС зависит от длительности и характеристики дебюта заболевания. Так, например, лечение IgA-НП до сих пор остается предметом дискуссий, в частности в отношении показаний к применению и выбору иммуносупрессивных препаратов [2, 4-6]. Как и при других ГЗ, при вариантах IgA-НП с активностью воспалительного процесса и нефротической протеинурией применяется сочетание цитостатиков с глюкокортикостероидами (ГКС) [7]. В последние годы при IgA-НП стал исследоваться препарат с селективным иммуносупрессивным действием - мофетила микофенолат (ММФ) [8].
При любом клиническом варианте, включая нефритический и нефротический синдромы, можно выявить МПГН - тяжелое хроническое заболевание почек, ведущее к терминальной почечной недостаточности. МПГН может быть идиопатическим, но чаще бывает вторичным на фоне хронических инфекций и системных иммунноопосредованных заболеваний [9, 10]. В развитых странах МПГН в последние десятилетия встречается все реже - в 1,8 % всех биопсий почки и в 3,7 % всех первичных ГН, что связано с успешной борьбой с инфекционными заболеваниями, включая вирусные гепатиты В и С.
В детском возрасте нередко встречаются состояния, связанные с генетическими мутациями, врожденными аномалиями развития почек. К ГЗ в данном случае относятся аномалии гломерулярной базальной мембраны (ГБМ) - наследственный нефрит, или синдром Альпорта, и болезнь тонких базальных мембран. Клинически они обычно характеризуются гематурией и/или умеренной протеинурией, хотя могут протекать и с НиС. Диагноз основывается на выявлении основных признаков болезни, анализе родословной ребенка, обследовании ближайших родственников [11].
Материал и методы
Исследование проводилось на базе нефрологического отделения Республиканской детской клинической больницы “Аксай” (Алматы). С помощью чрескожной пункционной биопсии почки, проведенной биопсийным пистолетом Gallini (Италия), под УЗИ-контролем проводился забор 2-3 столбиков коркового слоя почки. Морфологическое исследование почечного биоптата включало три необходимых исследования: световую (СМ), иммунофлюоресцентную (ИФМ) и электронную микроскопии (ЭМ), которые проводились нефропатологами А.В. Сухановым (Москва) и О.А. Воробьевой (Санкт-Петербург).
Диагноз НиС устанавливался на основании наличия у пациентов протеинурии, микро- или макрогематурии, отеков, снижения скорости клубочковой фильтрации (СКФ), артериальной гипертензии. В течение более 3 месяцев мы не включали в исследование пациентов с острым НиС. В основную группу вошли пациенты с морфологически верифицированным диагнозом НиС (27), в контрольную группу - пациенты с НиС без проведения биопсии (15). Длительность НиС составила в среднем 23,4 ± 5,2 месяца в основной группе и 25,1 ± 7,4 - в контрольной. Среднее артериальное давление (САД) у детей основной группы составило 132/74 мм рт. ст., протеинурия - в среднем 1,4 ± 1,7 г/с, макрогематурия в дебюте отмечена у 75 %, СКФ составила в среднем 67,5 ± 8,4 мл/мин. Среди детей контрольной группы САД составило 130/78 мм рт. ст., средние показатели протеинурии - 2,3 ± 1,5 г/с, СКФ - 68,5 ± 10,1 мл/мин, макрогематурия в дебюте имела место у 68 %. НиС часто (67,5 %) дебютировал на фоне или после купирования симптомов острой респираторной инфекции. Анамнез, отягощенный болезнями почек, отмечен у 2 (7,4 %) детей основной группы и у 2 (13,3 %) детей контрольной в виде ГН неясной этиологии (3) и хронической почечной недостаточности (1).
Результаты
Наиболее частым морфологическим вариантом НиС была IgA-НП - почти у % (74,1 % - 20, p < 0,001) детей. Раньше без проведения биопсии почки весь контингент пациентов с перечисленными заболеваниями лечился одинаково или неправильно. Среди 14,8 % (4) пациентов НиС протекал на фоне наследственного нефрита. Более редкими морфологическими вариантами были МПГН - 3,7 % (1), экстракапиллярный ГН - 3,7 % (1) и неуточненный муколипидоз - 3,7 %.
Для 65 % (13) из 20 детей IgA-НП была изолированной, т. е. ограниченной только почками, тогда как у остальных 35 % (7) детей IgA-НП носила системный характер, т. е. манифестировала на фоне пурпуры Шенлейн-Геноха в сочетании с кожным, абдоминальным и суставным синдромами.
При морфологическом исследовании, согласно классификации IgA-НП по Haas [12], у 7 детей имел место II класс, у 11 - III и у 2 - сочетание II и III классов. Острое повреждение канальцев было у 25 % (5) детей, инфильтрация интерстиция мононуклеарами - у 35 % (7). Эритроциты и эритроцитарные цилиндры в просвете канальцев отмечены для 35 % (7). Хронические изменения в виде фокально-диффузной атрофии канальцев, фиброза интерстиция возникли почти у половины детей - 45 % (9). Изменения в артериолах и мелких артериях в виде гипертрофии мышечного слоя и гиалиноза имели место у 35 % (7) детей. При ИФМ, конечно же, у всех детей установлено умеренное крупногранулярное свечение IgA в мезангии, более выраженное - при III классе. Кроме того, локализации свечения IgA у всех детей соответствовало таковое IgG и C3.
У 4 из 27 пациентов НиС протекал на фоне наследственного нефрита, аномалии ГБМ наследственного характера. Четкая отягощенная наследственность имела место у одной девочки.
На СМ у девочки 3,5 лет патологических изменений установлено не было. У остальных детей отмечен тотальный склероз 12-40 % всех клубочков, представленных в материале. У одного ребенка обнаружен слоистый вид ГБМ, ее утолщение. У троих детей отмечены диффузный фиброз интерстиция, а также диффузная атрофия канальцев. Стенки артерий и артериол были также утолщены за счет интимального фиброза у этих троих детей. У одного ребенка обнаружены многочисленные эритроцитарные цилиндры в канальцах. При ИФМ у всех детей отсутствовало свечение со всеми реагентами. ЭМ позволила установить характерные для наследственного нефрита аномалии ГБМ. ГБМ у всех детей была нерегулярной толщины с полной потерей нормальной трехслойной структуры, имея вид “баскетбольной корзины” с зазубренностью с эпителиальной стороны.
Для троих детей характерны другие морфологические варианты, лежащие в основе НиС. Так, неожиданной находкой для нас было обнаружение у девочки 12 лет признаков малоиммунного экстракапиллярного ГН с фиброзными и фиброзно-клеточными полулуниями в половине всех клубочков и полным склерозом 18 % клубочков, умеренным артериолосклерозом, мелкоочаговой атрофией канальцев и диффузным интерстициальным фиброзом. При ИФМ у девочки была умеренно выраженная экспрессия фибрина, свидетельствующая о повреждении целостности капилляров клубочков с выходом плазмы крови в мочевое пространство. У другой девочки, 12 лет, с МПГН II типа в дебюте была макрогематурия, перешедшая затем в персистирующую микрогематурию.
При морфологическом исследовании у ребенка обнаружены все характерные признаки МПГН по СМ в виде увеличения размеров клубочков, лобулярности, мезангиальной и эндокапиллярной пролиферации и выраженного утолщения стенки капилляров за счет мезангиальной интерпозиции. При ИФМ установлено умеренное свечение IgG и C3 вдоль стенки капилляров крупногранулярного характера, а также в мезангии. При ЭМ были установлены непрерывные электронно-плотные депозиты в толще ГБМ в виде “шнуров” диффузного характера в дополнение к подтверждению изменений, видимых при СМ.
Также необычной для нас находкой явилось установление у 4-летней девочки морфологических признаков тезаурисмоза, болезни накопления (неуточненной). При СМ в биоптате имели место увеличение клубочков в размерах, диффузные выраженные изменения подоцитов - гипертрофия и мелкокапельная вакуолизация с небольшим количеством неравномерного PAS-позитивного мелкогранулярного материала; в меньшей степени эти изменения фокально сегментарно были представлены в цитоплазме мезангиоцитов и париетальных эпителиальных клеток. В 20 % клубочков возникли явления позднего сегментарного гломерулосклероза. Изменения, подобные таковым подоцитов, имели место в цитоплазме эпителия извитых канальцев и интерстициальных макрофагов. При ИФ со всеми реагентами результат был отрицательным. При ЭМ цитоплазма подоцитов, мезангиоцитов, эпителия канальцев и интерстициальных клеток изобиловала оптически пустыми вакуолями с немногочисленными мембранными и слоистыми включениями. Не типичным для данного клинического случая был факт отсутствия неврологической симптоматики. Клинические особенности в зависимости от морфологического варианта НиС представлены в таблице.
Таблица.Клинические характеристики и морфологические варианты нефритического синдрома у детей (n = 27).
Примечание. МПГН — мембрано-пролиферативный гломерулонефрит; ЭКГН- экстракапиллярный гломерулонефрит; АД — артериальное давление; АГ — артериальная гипертензия; ГУ — гематурия.
* Достоверное различие между группами, p < 0,05.
Для индукции ремиссии НиС применялись пульсовое внутривенное введение метилпреднизолона в течение нескольких дней, пероральный преднизолон (ПЗ) 60 мг/м2/сут, ММФ (СеллСепт®, Hoffmann-La Rosh) 1 г/м2/с в течение 3-4 месяцев. При достижении ремиссии, что устанавливалось по купированию отеков, артериальной гипертензии, уменьшению протеинурии до < 0,5 г/с, назначалась поддерживающая терапия: ПЗ 40 мг/м2/48 ч со снижением дозы (коротким курсом), ММФ 500 мг/м2/с год и более. Все дети тоже получали ингибиторы АПФ с антипротеинурической и нефропротективной целью (фозиноприл, эналаприл в дозе 2,5-20 мг в зависимости от возраста, наличия артериальной гипертензии). Больные контрольной группы получали циклофосфамид (ЦФ) в виде 3-4 внутривенных пульсов (20 мг/кг/3-4 недели) либо перорально 2-2,5 мг/кг/с 2 месяца в сочетании с ПЗ 60 мг/м2/ сут в течение 1,5—2,0 месяцев и последующим переходом на альтернирующий режим 40 мг/м2/48 ч со снижением. Все они также получали иАПФ.
Сравнение эффективности лечения пациентов основной группы с IgA-НП и пациентов контрольной группы представлено на рис. 1.
Рисунок 1. Динамика протеинурии (г/сут) у детей с нефритическим синдромом на фоне иммуносупрессивной терапии.
При сравнении протеинурии в конце катамнеза нами установлена достоверно (р < 0,05) меньшая протеинурия у пациентов основной группы по сравнению с контрольной.
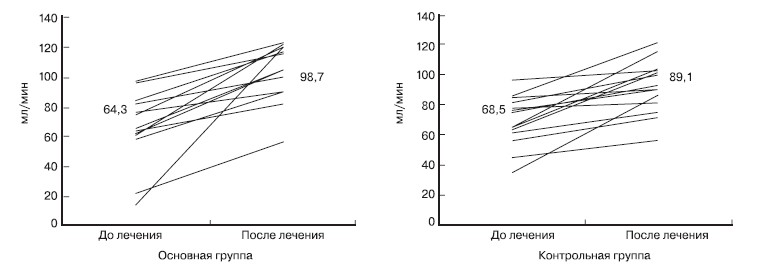
Оценка динамики СКФ на фоне лечения в обеих группах представлена на рис. 2.
Рисунок 2. Динамика скорости клубочковой фильтрации (мл/мин) у детей с нефритическим синдромом на фоне иммуносупрессивной терапии.
У детей основной группы на фоне лечения отмечено повышение СКФ с 64,3 ± 23,5 до нормы - 98,7 ± 21,5 мл/мин, тогда как у детей контрольной группы исходная СКФ 68,5 ± 10,1 мл/ мин нормального значения не достигала - 89,1 ± 21,4 мл/мин. Среди пациентов основной группы с IgA-НП оказалась высокой эффективность ММФ в отношении снижения уровня протеинурии и с увеличением длительности лечения степень снижения протеинурии увеличивалась. Так, в группе детей с IgA-НП, исходная протеинурия 2,3 ± 0,4 г/с уже 6 шесть месяцев лечения снизилась до 1,7 ± 0,2 г/с; а через 2 года - почти до следов - 0,14 ± 0,03 г/с (р < 0,01). Среди пациентов контрольной группы с НиС без биопсии, получавших ЦФ, также имело место снижение исходной протеинурии - с 2,5 ± 1,5 г/с до 0,8 ± 0,3 г/с в первые полгода с повышением до 2,2 ± 0,4 г/с в течение второго года. Дальнейшего снижения протеинурии после первого года лечения не отмечено.
На фоне лечения ЦФ возникли побочные эффекты, из которых наиболее частыми были инфекционные осложнения, отмечавшиеся почти у половины (46,7 % - 7) пациентов. Для одной пятой (20 % - 3) пациентов оказались характерными токсический гепатит и диспепсические явления, реже - алопеция (6,7 % - 1). На фоне лечения ММФ в основной группе имели место только частые инфекции (14,3 % - 3), а описываемые в литературе расстройства пищеварения, связанные с местным токсическим воздействием ММФ, или анемия не встречались. Возможно, это связано с тем, что описанные в литературе побочные эффекты наблюдались среди пациентов на более высоких дозах ММФ после трансплантации донорской почки.
Обсуждение
Таким образом, для наших пациентов НиС был обусловлен преимущественно IgA-НП. Терапия ММФ в сочетании с ГКС и иАПФ при IgA-НП с НиС эффективна и более безопасна по сравнению с пациентами контрольной группы, получавшими лечение ЦФ без морфологического подтверждения диагноза. К сожалению, до сих пор нет метода лечения, влияющего на депозиты IgA в мезангии. Имеющиеся методы лечения в основном направлены на иммунный и воспалительный процессы в клубочках и тубулоинтерстиции, ведущие к склерозированию почечной ткани. Требуются многолетние контролируемые рандомизированные исследования на большой группе пациентов для оценки различных протоколов иммуносупрессивной терапии при IgA-НП [13]. Тем не менее впервые представляемый нами опыт лечения IgA-НП в Казахстане показывает целесообразность проведения патогенетической иммуносупрессивной терапии, позволяющей улучшать течение и прогноз заболевания у пациентов с IgA-НП.
Необычным оказался тот факт, что НиС протекал на фоне аномалий ГБМ у 4 из 27 пациентов НиС. При морфологическом исследовании у большинства детей имели место тотальный склероз до 40 % всех клубочков, представленных в материале, типичный вид ГБМ и диффузный фиброз интерстиция, атрофия канальцев, тогда как иммунофлюоресценция для всех детей была отрицательной. Неожиданной находкой для нас было обнаружение малоиммунного ЭКГН, тезаурисмоза и МПГН II типа. Эти дети получали только иАПФ с хорошим эффектом по снижению протеинурии и повышению СКФ.



{kind=link}
{kind=link}
{kind=link}