
Введение
Атипичный гемолитико-уремический синдром (аГУС) – системное заболевание группы тромботических микроангиопатий (ТМА), обусловленное неконтролируемой активацией альтернативного пути комплемента вследствие генетического дефекта (мутации генов, кодирующих синтез белков – регуляторов системы комплемента) или реже – аутоиммунной патологии (выработка аутоантител к фактору Н – основному регуляторному протеину альтернативного пути) [1, 2]. Имеющиеся у пациентов с аГУС мутации в генах факторов Н (CFH), I (CFI), мембранного кофакторного протеина (MCP), тромбомодулина (THBD) обусловливают их количественный или чаще функциональный дефицит, что приводит к нарушению защиты эндотелия от атаки комплемента. Результатом этого становится усиленное образование на поверхности клеток мембрано-атакующего комплекса (МАК), вызывающего их повреждение с обнажением субэндотелиального матрикса, трансформацию атромботического фенотипа в протромботический, активацию тромбоцитов с последующим тромбообразованием в сосудах микроциркуляторного русла различных органов [3, 4]. Таким образом, аГУС представляет собой комплемент-опосредованную ТМА, характеризующуюся классической триадой симптомов – микроангиопатической гемолитической анемией (МАГА), тромбоцитопенией и острым почечным повреждением, поскольку именно почки служат основной (однако не единственной) мишенью микротромбообразования [5]. Атипичный ГУС имеет крайне высокий риск внезапной смерти и необратимых инвалидизирующих повреждений жизненно важных органов, включая почки, печень, сердце и головной мозг [6]. Хотя комплемент-опосредованная ТМА поражает главным образом сосуды почечной микроциркуляции, в последнее время все чаще отмечают экстраренальные проявления аГУС, которые развиваются не менее чем у 20% пациентов. Помимо упомянутых выше у больных аГУС описаны также поражения легких, желудочно-кишечного тракта, кожи, скелетных мышц, глаз. Такой широкий спектр экстраренальных проявлений, безусловно, свидетельствует об истинно системном характере комплемент-опосредованной ТМА. А тот факт, что эти проявления могут развиваться не только в момент острого эпизода ТМА, но и спустя несколько лет после его купирования, а также у пациентов, получающих диализную терапию (у которых, как полагали ранее, невозможно развитие новых эпизодов ТМА), лишь подтверждает представление о возможности хронического течения аГУС, обусловленного постоянно происходящей активацией комплемента, с вовлечением в патологический процесс новых бассейнов микроциркуляции [7, 8]. Полагают, что генерализация микроангиопатического процесса может быть связана с воздействием на организм все увеличивающегося числа различных неблагоприятных факторов, потенциально способных вызывать дополнительное повреждение эндотелия у лиц, имеющих генетический дефект регуляции системы комплемента (воспаление, лекарства, радиация, артериальная гипертензия и т.п.) [7].
Из всех экстраренальных проявлений аГУС наибольшее значение имеют поражение ЦНС и сердца, поскольку их патология может быть ассоциирована с неблагоприятным прогнозом. Точная распространенность поражения сердца и степень его влияния на общий прогноз при аГУС не известны, что обусловлено отсутствием характерной клинической картины и, следовательно, общепринятых критериев обследования данных пациентов. Между тем есть основания полагать, что при аГУС, как и при любой ТМА, возможно поражение сердца различной степени тяжести, вплоть до развития некоронарогенного инфаркта миокарда [9–11], обусловленного тромбозами мелких интрамиокардиальных сосудов. В настоящей статье мы хотели бы продемонстрировать различные варианты нарушения сердечной функции у пациентов с аГУС.
Пациенты и методы
Нами были проанализированы клинические проявления 17 больных аГУС, наблюдавшихся в клинике нефрологии им. Е.М. Тареева Первого МГМУ им. И.М. Сеченова и нефрологическом отделении ГКБ им. А.К. Ерамишанцева Москвы в 2010–2013 гг. У 5 (29,4%) из них выявлены различные документально подтвержденные признаки поражения сердца. Приводим наши наблюдения.
Пациент 1 18 лет. Летом 2012 г. после введения противостолбнячной сыворотки развился нефротический синдром (НС). Проводимая в течение 3 месяцев иммуносупрессивная и диуретическая терапия оказалась неэффективной, отеки быстро наросли до степени анасарки, в связи с чем в октябре 2012 г. был госпитализирован в нефрологическую клинику Первого МГМУ им. И.М. Сеченова. При обследовании картина развернутого НС (суточная протеинурия (СПУ) 8 г/сут, альбумин крови 22 г/л), олигурия, анасарка, креатинин крови 1,2 мг/дл, СКФ 61 мл/мин. Обращала на себя внимание анемия (гемоглобин 119 г/л), повышение уровня ЛДГ (990 ЕД/л). В связи с резистентностью отеков к массивной диуретической терапии проведено 13 сеансов ультрафильтрации, в результате чего отеки уменьшились, вес пациента снизился на 15 кг. Однако выраженность НС нарастала (СПУ 13 г/сут, альбумин крови 17 г/л), отмечено снижение гемоглобина до 89 г/л, повышение уровня ЛДГ до 1300 ЕД/л, креатинина – до 2,0 мг/дл.
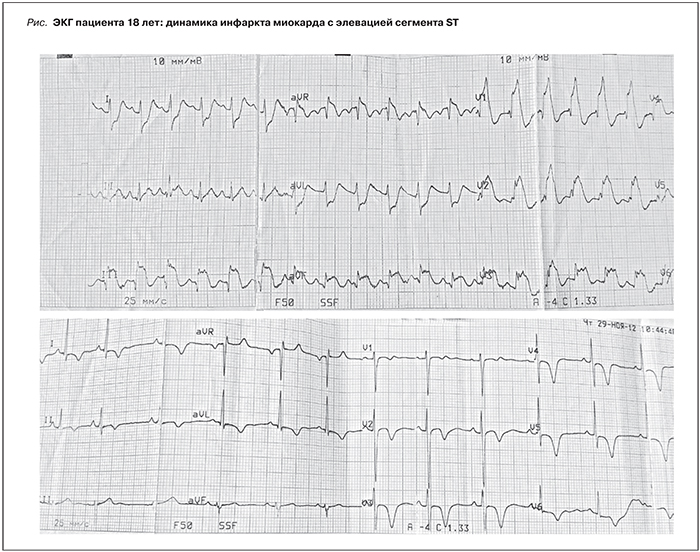
Через 3 недели от момента госпитализации – эпизод потери сознания с агональным дыханием. Констатирована клиническая смерть, начаты реанимационные мероприятия. Через 3–5 минут отмечено восстановление дыхания и сердечно-сосудистой деятельности, был переведен в ОРиИТ в крайне тяжелом состоянии. На ЭКГ – признаки острого переднего с циркулярным поражением верхушки подъемом сегмента ST инфаркта миокарда (элевация ST III, aVF, V1–V5) (см. рисунок), диагностически значимым повышением тропонина I (0–1,3–0,6 нг/мл). При этом какие-либо кардиологические жалобы отсутствовали. В дальнейшем – рецидивирующая левожелудочковая недостаточность. Проведенная коронароангиография (КАГ) исключила патологию коронарных артерий, что позволило диагностировать некоронарогенный инфаркт миокарда. С целью исключения ТЭЛА была также проведена МСКТ органов грудной клетки, не выявившая патологических изменений. При МРТ головного мозга обнаружены множественные очаги ишемии, расцененные как проявления нарушения мозгового кровообращения в бассейнах терминальных ветвей правой и левой передних мозговых артерий и центральных ветвей левой задней мозговой артерии по ишемическому типу.
Для уточнения причины сердечно-сосудистой катастрофы с развитием полиорганной недостаточности было предпринято дополнительное обследование молодого пациента. Уровень ADAMTS-13 составил 98% (норма), выявлено снижение в крови активности всех компонентов комплемента, скринирующее исследование обнаружило возможный дефицит фактора Н. Полученные данные в сочетании с микроангиопатическим гемолизом, умеренной транзиторной тромбоцитопенией и ОПН позволили диагностировать атипичный ГУС, осложнивший течение ХГН нефротического типа. Была начата плазмотерапия (инфузии свежезамороженной плазмы), которая сочеталась с длительным непрерывным внутривенным введением гепарина (через инфузомат 12 500 ЕД/сут) и метилпреднизолона 250 мг/сут в/в капельно. В результате лечения состояние пациента стабилизировалось. Признаки микроангиопатического гемолиза купированы (гемоглобин – 113 г/л, ЛДГ – 546 ЕД/л), функция почек восстановилась (креатинин крови – 0,69 мг/дл, СКФ – 124 мл/мин). По данным Эхо-КГ нарушения локальной и глобальной сократимости миокарда ЛЖ не выявлено. Однако на ЭКГ отмечена динамика, характерная для инфаркта миокарда с формированием отрицательных зубцов T в грудных отведениях (V2–V6) (см. рисунок).
Пациент 2 25 лет. Дебют заболевания за 2 года до поступления в клинику в виде развития остронефритического синдрома, острой почечной недостаточности (ОПН) (макрогематурия, отеки, олигурия, артериальная гипертензия – 170/110 мм рт.ст., креатинин – 4,2 мг/дл). Кардиологические жалобы отсутствовали. При обследовании в стационаре по месту жительства выявлены снижение уровня гемоглобина до 67 г/л, тромбоцитопения до 28 тыс. в 1 мкл, повышение уровня ЛДГ до 900 ЕД/л. Проведена антибактериальная, инфузионная, диуретическая терапия без эффекта в отношении восстановления функции почек. Почечная недостаточность нарастала, развилась анурия, в связи с чем начаты сеансы гемодиализа. С целью уточнения диагноза проведена биопсия почки, выявившая характерную картину острой ТМА. Диагностирован аГУС. Через 3 месяца после дебюта заболевания при сохраняющихся лабораторных маркерах ТМА выявлены признаки поражения сердца по данным Эхо-КГ: умеренная дилатация левого предсердия (5,4×4,4 см), снижение глобальной сократимости ЛЖ – ФВ 40% (биплановым методом), гипокинез заднего отдела МЖП, базальных и средних сегментов задней и задне-боковой стенок ЛЖ. ЭКГ без значимой патологии. Наблюдались рецидивирующие тромбозы в местах постановки центрального венозного катетера (левых брахиоцефальной и подключичной вен), а также правых подвздошной и бедренной вен, в связи с чем был установлен кава-фильтр. Для уточнения причины рецидивирующего венозного тромбообразования, не характерного для аГУС, исследован полиморфизм генов гемостаза, выявлены признаки наследственной тромбофилии (гомозиготная мутация в гене метионин-синтазы, гетерозиготные мутации в генах ингибитора активатора плазминогена, фибриногена, тромбоцитарного гликопротеина 1В, коагуляционного фактора XII). С учетом выявленных изменений проведено интенсивное лечение: 10 сеансов плазмообмена, антикоагулянтная терапия (варфарин с целевыми значениями МНО 3,0–3,5). Однако ОПН не разрешилась, в связи с развитием терминальной ХПН продолжено лечение программным гемодиализом. При контрольной Эхо-КГ через 1 месяц полости сердца не расширены, ФВ 64% (Тейхольц), асинхронное движение МЖП с участками гипокинезии. Через 2 года после острого эпизода в связи с жалобами на одышку проведена перфузионная томосцинтиграфия миокарда (ОЭКТ, исследование в покое, индикатор 99 мТс-МИБИ с шагом томографии 6,75 мм), нарушений в распределении индикатора не выявлено.
Пациент 3 29 лет. Дебют заболевания в возрасте 28 лет, когда во время приступа головных болей впервые отмечено повышение АД до 200/110 мм рт.ст. За медицинской помощью не обращался, нерегулярно принимал энап. Через полтора года существования артериальной гипертензии после пребывания в жарком климате появился навязчивый сухой кашель, одышка в покое. При обращении в поликлинику кроме АД 200/120 мм рт.ст. выявлены анемия (гемоглобин 104 г/л) и выраженная азотемия (креатинин крови 6,7 мг/дл). Госпитализирован в нефрологический стационар, где в ходе обследования отмечены нарастание анемии (Нв 91 г/л), снижение количества тромбоцитов в динамике (в течение 1 месяца с 319 до 153 тыс. в 1 мкл), повышение уровня ЛДГ до 724 ЕД/л, минимальная протеинурия (0,25 г/л) без изменения мочевого осадка при сохранном диурезе (1500 мл/сут), дальнейшее нарастание уровня креатинина (7,5 мг/дл). При ЭХО-КГ выявлены расширение полостей левого желудочка и левого предсердия, диффузный гипокинез и умеренная гипертрофия миокарда ЛЖ, фракция выброса 43% (Тейхольц), утолщение створок митрального клапана, признаки легочной гипертензии (повышение СДЛА до 51 мм рт.ст.), небольшое количество жидкости в полости перикарда. ЭКГ без значимой патологии. Консультирован кардиологом, установившим диагноз заднедиафрагмального ИМ неизвестной давности. Для определения причины быстронарастающей почечной недостаточности, тяжелой артериальной гипертензии и перенесенного ИМ в отношении молодого человека предпринято обследование, исключившее системные заболевания (системную красную волчанку [СКВ], АНЦА-ассоциированные васкулиты, антифосфолипидный синдром [АФС], реноваскулярную гипертензию. С целью уточнения характера поражения почек выполнена пункционная биопсия, выявившая морфологическую картину хронической ТМА: В биоптате 22 клубочка, клубочки стянуты друг к другу, 2 из них полностью склерозированы, в 4 – ишемия капиллярных петель, еще в одном клубочке капиллярные петли коллабированы, стенка их утолщена, имеются двойные контуры. Диффузный склероз интерстиция, атрофия канальцев, занимающая более 70% площади паренхимы. Неспецифическая инфильтрация интерстиция воспалительными клетками в зоне склероза. В двух артериях малого калибра – миоинтимальная пролиферация и склероз интимы; в артериолах – мукоидное набухание интимы с сужением просвета сосудов. При ИФ специфического свечения не обнаружено.
Принимая во внимание особенности клинико-лабораторной картины и течения заболевания, данные гистологического исследования ткани почки, отрицательные серологические маркеры АФС, установлен диагноз аГУС. Обращала на себя внимание выраженная активация внутрисосудистого свертывания крови (уровень растворимых комплексов фибрин-мономеров [РКФМ]повышен в 4 раза [норма до 4 мг/дл]), в связи с чем было выполнено исследование полиморфизма генов гемостаза, выявившее мультигенную тромбофилию, представленную гомозиготными мутациями в генах метионинсинтазы-редуктазы и фибриногена, гетерозиготными – в генах ингибитора активатора плазминогена и тромбоцитарного гликопротеина 1В. Было высказано предположение о том, что гиперкоагуляционные изменения системы гемостаза могли усилить тромбообразование в сосудах микроциркуляторного русла почек и сердца, что послужило основанием для назначения низкомолекулярного гепарина (клексан 80 мг/сут п/к) в дополнение к терапии конкором, энапом и тромбо-ассом. Через месяц от начала терапии состояние пациента улучшилось, исчезла одышка, уровень креатинина снизился до 5,0 мг/дл, АД стабилизировалось на цифрах 130/90 мм рт.ст., нормализовались гемоглобин и количество тромбоцитов. При контрольном Эхо-КГ через месяц выявлены гипо-акинез задне-нижней, нижней, нижне-перегородочной стенок ЛЖ, повышение СДЛА до 53 мм рт.ст. ФВ возросла до 50% (Тейхольц). В дальнейшем антикоагулянтная терапия продолжена, пациент в течение полутора лет непрерывно получал фраксипарин 0,3 мл/сут, что позволило стабилизировать функцию почек (креатинин крови 4 мг/дл, СКФ 28–30 мл/мин), ФВ нормализовалась – 56%.
Пациент 4 30 лет. Заболел остро в конце мая 2013 г., когда без видимой причины появились интенсивные боли в эпигастрии, продолжавшиеся в течение 4 дней. Бригадой СМП зафиксировано повышение артериального давления до 170/100 мм рт.ст., элевация сегмента ST, в связи с чем был госпитализирован в ОРИТ с диагнозом «нестабильная стенокардия. Острый панкреатит». В ходе обследования выявлены анемия (Нв 78 г/л) с признаками микроангиопатического гемолиза (уровень ЛДГ – 1116 ЕД/л), тромбоцитопения (число тромбоцитов – 85 тыс. в 1 мкл), быстронарастающая почечная недостаточность (креатинин крови в динамике – 445-618-759-989 мкмоль/л). ЭКГ без значимой патологии. Проведенная Эхо-КГ выявила дилатацию левого предсердия, выраженную концентрическую гипертрофию и диффузный гипокинез миокарда ЛЖ с умеренным снижением его глобальной сократимости до ФВ 49% (Тейхольц). В связи с прогрессированием почечной недостаточности пациент был переведен в нефрологическое отделение ГКБ им. А.К. Ерамишанцева. В тот момент тяжесть состояния была обусловлена нарастанием азотемии (креатинин – 1057 мкмоль/л), тяжелой артериальной гипертензией (АД 220/120 мм рт.ст.), резистентной к 4-компонентной антигипертензивной терапии, олигурией, гипергидратацией (двусторонний гидроторакс, асцит, периферические отеки), прогрессирующей анемией (Нв 75 г/л). Кроме того, персистировал интенсивный болевой синдром в эпигастрии и околопупочной области, в связи с чем пациенту выполнена диагностическая лапароскопия, позволившая отвергнуть острую хирургическую патологию. Проведенная ЭГДС выявила признаки эрозивно-язвенного гастродуоденита. В связи с нарастанием почечной недостаточности, гиперкалиемией, гипергидратацией, некорригируемой артериальной гипертензией 11 июля 2013 г. начато лечение программным гемодиализом.
С целью морфологической верификации характера поражения почек пациенту была выполнена нефробиопсия, выявившая характерную картину хронической тромботической микроангиопатии: в препарате 15 клубочков, один из них полностью склерозирован, в остальных ишемия капиллярных петель различной выраженности. Диффузный склероз интерстиция и атрофия канальцев, занимающие более 70% площади паренхимы. Неспецифическая плотная инфильтрация интерстиция воспалительными клетками в зонах склероза. Артерии: имеется несколько профилей артерий среднего калибра, просвет которых резко сужен вплоть до полной окклюзии за счет расширения субэндотелиального пространства, мукоидного набухания и склероза интимы по типу «луковой шелухи», артериолы – артериолосклероз. При иммунофлюоресцентном исследовании – отсутствие свечения всех иммуноглобулинов, компонентов комплемента, фибриногена, легких цепей.
Дальнейшее обследование было направлено на уточнение нозологической формы ТМА. Были обнаружены нормальная активность ADAMTS-13 (112%), что позволило исключить тромботическую тромбоцитопеническую пурпуру (ТТП), снижение уровня С3 компонента комплемента в отсутствие серологических маркеров СКВ и АФС. С учетом характера течения заболевания, признаков полиорганного поражения (почки, сердце, ЖКТ), морфологического исследования ткани почки, результатов анализов диагностирован атипичный ГУС.
Было начато лечение инфузиями свежезамороженной плазмы, которое, однако, пришлось быстро отменить из-за плохой переносимости низкомолекулярным гепарином (Клексан 60 мг/сут п/к), продолжена многокомпонентная антигипертензивная терапия. В течение месяца состояние больного стабилизировалось, число тромбоцитов и уровень ЛДГ нормализовались, выраженность анемии уменьшилась. Однако функция почек не восстановилась, пациент остался диализзависимым, сохранялась также артериальная гипертензия, требующая медикаментозного лечения.
Пациентка 5 30 лет. Заболела остро в сентябре 2013 г. на сроке 23–24-й недели беременности, когда появились многократный жидкий стул (6–8 раз в сутки), ломота во всем теле, боли в мышцах, олигурия, учащенное сердцебиение, кожный зуд. Была госпитализирована в ИБ № 1, откуда в связи с развитием острой почечной недостаточности переведена в ОРИТ ГКБ им. А.К. Ерамишанцева. В момент поступления тяжесть состояния определена выраженной анемией (Нв 66 г/л) с признаками микроангиопатического гемолиза (ЛДГ – 843 ЕД/л), тромбоцитопенией (56 тыс. в 1 мкл), острой почечной недостаточностью (анурия, гипергидратация, креатинин крови 577 мкмоль/л), потребовавшей начала заместительной почечной терапии сеансами продленной вено-венозной гемодиафильтрации (ПВВГДФ) на аппарате PRISMA, артериальной гипертензией (АД 150/100 мм рт.ст.), сердечной недостаточностью (при ЭХО-КГ диффузное снижение глобальной сократимости миокарда ЛЖ: ФВ 39% [Тейхольц], жидкость в полости перикарда, дилатация обоих предсердий, недостаточность трикуспидального клапана). В течение нескольких дней присоединились признаки дыхательной недостаточности (рецидивирующий отек легких, двусторонняя полисегментарная пневмония), что потребовало начала ИВЛ с последующей трахеостомией. Несмотря на попытки пролонгирования беременности, у пациентки развилась преэклампсия, потребовавшая досрочного оперативного родоразрешения 03.10.2013, ребенок умер на 2-е сутки. C учетом острого развития полиорганной недостаточности у беременной женщины с признаками микроангиопатического гемолиза и тромбоцитопении диагноз ТМА сомнений не вызывал. Срок беременности, при котором возникла ургентная ситуация, требовал проведения дифференциальной диагностики между ТТП и аГУС. С этой целью выполнено исследование ADAMTS-13, уровень которого составил 60% (референсные значения – 80–113%), что позволило исключить ТТП. Установлен диагноз аГУС. Начата интенсивная плазмотерапия в режимах плазмообмена или инфузий свежезамороженной плазмы ежедневно в сочетании с антикоагулянтами (клексан – 60 мг/сут), антибактериальная терапия, дважды проводились трансфузии эритроцитной массы, продолжена ПВВГДФ. В течение 8 дней больная оставалась на ИВЛ.
В результате лечения состояние больной стабилизировалось, к концу 3-й недели терапии отмечено полное восстановление функции почек (креатинин 71 мкмоль/л, рСКФ 124 мл/мин (CKD-EPI), явления сердечной недостаточности были полностью купированы (ФВ – 60%), АД и число тромбоцитов нормализовались.
Особенности поражения сердца при тромботических микроангиопатиях
Поражение сердца возможно при любых ТМА. Впервые оно было описано еще в 1925 г. американским клиницистом и патологом Э. Мошковицем у 16-летней девушки с ТТП, которая помимо гемолитической анемии и тромбоцитопении проявлялась поражением ЦНС, сердца в виде острой левожелудочковой недостаточности и почек [12]. Через 6 дней пациентка умерла от полиорганной недостаточности, гистологическое исследование обнаружило распространенный тромбоз терминальных артериол и капилляров головного мозга, сердца и почек. К настоящему времени известно множество случаев развития у больных ТТП сердечной недостаточности и острого инфаркта миокарда [11]. Сегодня сердце рассматривается как один из основных наряду с головным мозгом органов-мишеней микроциркуляторного тромбообразования при ТТП, а его поражение – как неблагоприятный прогностический фактор и важная причина смерти у этого контингента больных [9, 11]. При аутопсийном исследовании в подавляющем большинстве случаев выявляют тромбоз интрамиокардиальных артериол и капилляров и очаги некроза миокарда в отсутствие патологии коронарных артерий, реже – кровоизлияния в миокард и очаговый фиброз миокарда [11, 13, 14].
Длительное время считалось, что микроангиопатическое поражение миокарда возможно лишь у больных ТТП, однако оказалось, что ТМА миокарда развивается также при типичном и атипичном ГУС, причем не только у взрослых пациентов, но и у детей [7, 8, 15]. По современным представлениям, острые или хронические сердечно-сосудистые проявления регистрируются у 3–10% пациентов с аГУС. Особой предрасположенностью к поражению сердца отличаются больные с мутациями факторов CFH и CFВ-комплемента [8, 16]. Поражение сердца наиболее часто представлено при аГУС инфарктом миокарда, кардиомиопатией и сердечной недостаточностью [3, 7, 8, 10]. Однако, как и при ТТП, описаны выпот в полость перикарда с тампонадой сердца, нарушения ритма и проводимости, кардиогенный шок, синкопальные состояния, не связанные с поражением легких и ЦНС, а также внезапная смерть [9, 10, 13]. Последняя, по-видимому, также может быть обусловлена аритмиями, поскольку при аутопсийном исследовании миокарда умерших пациентов с ТТП был выявлен тромбоз сосудов в области атриовентрикулярного узла и пучка Гиса [11, 17, 18]. Морфологическая картина поражения миокарда при обоих заболеваниях также не различается на светооптическом уровне: гистопатологическое исследование выявляет очаги некроза миокарда, утолщение стенок мелких сосудов, отек эндотелиальных клеток, расширение субэндотелия, интралюминальные тромбы. Однако при иммуногистохимическом исследовании обнаруживают экспрессию МАК в стенке сосудов микроциркуляторного русла миокарда и инфарцированных кардиомиоцитах, свидетельствующую об активации комплемента, при аГУС и депозиты фактора фон Виллебранда в просвете сосудов при ТТП [10, 19].
Механизмы развития ТМА, в т.ч. сосудов интрамиокардиальной микроциркуляции, не ограничиваются лишь прямым повреждением эндотелиальных клеток, опосредованным активацией комплемента или избыточным тромбообразованием, обусловленным накоплением сверхкрупных мультимеров фактора фон Виллебранда. Дополнительный вклад в развитие микроциркуляторных тромбозов вносит также повреждающее воздействие на эндотелий свободного гемоглобина, появляющегося в циркуляции в результате гемолиза. Как было установлено, свободный гемоглобин, и особенно гем, способны активировать эндотелиальные клетки, индуцируя воспаление и тромбоз, а также подавлять продукцию ими оксида азота (NO), приводя к вазоконстрикции и активации тромбоцитов [13, 20]. Кроме того, недавно в условиях in vitro было показано, что гемоглобин может ингибировать активность металлопротеазы ADAMTS 13, расщепляющей сверхкрупные мультимеры фактора фон Виллебранда что, по-видимому, также способствует усилению процесса микротромбообразования [21]. Представляется, что этот механизм может быть особенно важен для ТТП, развитие которой обусловлено выраженным дефицитом активности ADAMTS 13 (менее 5%). Нельзя исключить, что при имеющемся исходно дефиците активности фермента любой дополнительный фактор, усугубляющий его, может способствовать генерализации тромбообразования в мелких сосудах, формируя «системность», характерную для ТТП, в т.ч. и за счет поражения микроциркуляторного русла миокарда.
Обсуждение
Представленные наблюдения иллюстрируют возможность и особенности поражения сердца у пациентов с аГУС. Среди больных нашей когорты аГУС (всего 17) частота поражения сердца составила 29,4%, что почти втрое превышает данные других авторов [7, 8]. У всех представленных пациентов поражение сердца развилось во время острого эпизода ТМА и во всех случаях – в рамках полиорганного поражения. При этом все больные имели признаки острого почечного повреждения (ОПП) разной выраженности: от небольшого повышения креатинина крови у пациента 1 до тяжелой острой почечной недостаточности, потребовавшей применения гемодиализа (больные 2, 4, 5) и разрешившейся с полным восстановлением функции почек без дальнейшей потребности в диализной терапии только у больной 5. У пациентов 2 и 4 в исходе ОПП развилась терминальная почечная недостаточность, и они остались диализзависимыми, а у больного 3, который не нуждался в проведении гемодиализа в дебюте заболевания, несмотря на выраженное нарушение функции почек, сформировалась хроническая почечная недостаточность, в течение 1,5 лет не прогрессирующая. Принимая во внимание сочетание ОПП с острой сердечной недостаточностью у всех больных, мы можем констатировать развитие у них острого кардиоренального синдрома (КРС) 5-го типа (вторичного) – одновременного нарушения функции сердца и почек, наблюдающегося при различных заболеваниях, включая системные болезни, сепсис, сахарный диабет [22]. До настоящего времени ТМА не упоминались в качестве причины КРС. Однако, опираясь на пример наших пациентов и данные литературы [7, 8], мы полагаем, что любые системные ТМА, к которым относятся аГУС и ТТП, могут по праву считаться важными причинами развития острого КРС 5-го типа.
Следует отметить, что у всех наших пациентов с аГУС, кроме больного 2, острая сердечная недостаточность развивалась в дебюте болезни практически одновременно с ОПП при наличии тромбоцитопении и выраженных признаках микроангиопатического гемолиза, что согласуется с данными других авторов, отметивших этот факт как особенность аГУС преимущественно у пациентов с мутациями фактора Н или анти-CFH-антителами [8, 10, 16]. Результаты скрининга у пациента 1 в нашей небольшой когорте больных, позволяющие предполагать дефицит фактора Н, косвенно подтверждают наблюдения коллег. У пациента 2 в дебюте заболевания поражение сердца отсутствовало, однако его симптомы появились спустя 3 месяца, когда гематологические проявления были уже купированы, а больной получал лечение гемодиализом. Таким образом, наши данные согласуются с имеющимися в литературе сведениями о том, что развитие острой сердечной патологии может происходить и после наступления терминальной почечной недостаточности у пациентов, находящихся на гемодиализе, и значительно осложнять течение основного заболевания [8].
Обращает на себя внимание отсутствие типичной клинической картины поражения сердца у всех вышеописанных больных, что совпадает с результатами ранее проведенных исследований. По данным крупного когортного исследования, включившего 275 пациентов с ТТП и аГУС, боль в области сердца была описана лишь у 6% из них [23]. Однако, по данным аутопсий больных, умерших от ТТП, у 100% были выявлены участки некроза миокарда вследствие тромбоза мелких коронарных сосудов [17]. По-видимому, безболевой характер ишемии миокарда можно считать важной особенностью поражения сердца при ТМА разного генеза даже в случае развития инфаркта миокарда. Аргументом в пользу этого предположения служит пример пациента 1, у которого ОИМ манифестирован остановкой сердца в отсутствие предшествующего болевого синдрома. Аналогичная картина описана M. Sallee и соавт. у 43-летней женщины с аГУС, клинически проявившимся характерными гематологическими изменениями, ОПН и нефротическим синдромом. Уже после того, как микроангиопатический гемолиз и тромбоцитопения были купированы, она продемонстрировала внезапную остановку сердца. Несмотря на немедленно начатые реанимационные мероприятия, больная умерла. При аутопсийном исследовании, выполненном через 12 часов после смерти, диагностирован инфаркт миокарда в отсутствие признаков поражения коронарных артерий. Гистологическое исследование выявило множественные очаги некроза кардиомиоцитов в обоих желудочках давностью от 15 дней до 24 часов и характерные признаки ТМА в виде утолщения стенки мелких артерий и отека субэндотелия в отсутствие тромбоза [10].
Анализ симптомов поражения сердца у 5 представленных пациентов с аГУС выявил еще одну особенность – отсутствие значимых изменений ЭКГ у 4 из них, за исключением одного, у которого развился инфаркт миокарда. Единственным изменением ЭКГ, которое было зарегистрировано у всех наших больных в момент обследования, была синусовая тахикардия, которая отмечается и другими авторами как наиболее частое нарушение при ТМА [11, 23].
У упомянутых 4 пациентов, однако, имелись эхокардиографические данные, свидетельствующие о вовлечении в патологический процесс сердца. Во всех 4 случаях наблюдалось снижение глобальной сократимости миокарда ЛЖ (хотя бы незначительное: ФВ от 39 до 49%, в среднем 43%) на фоне диффузного или локального гипокинеза. У всех этих пациентов имело место расширение камер сердца, причем у трех выявлено расширение левых предсердий, а у пациентки 5 – в сочетании с расширением правого предсердия. Только одному пациенту была проведена КАГ, по данным которой было исключено поражение крупных коронарных артерий. Следует отметить, что сегодня это исследование вообще редко выполняют больным ТМА. Так, по данным B.M. Hawkins и соавт., лишь двоим из 111 пациентов с ТТП, имевших признаки поражения сердца, была проведена КАГ, причем в обоих случаях патология крупных коронарных артерий отсутствовала [11], как и в нашем наблюдении.
Учитывая молодой возраст представленных нами пациентов (до 30 лет) и отсутствие кардиологического анамнеза, можно с большой долей вероятности отрицать у них наличие классической ИБС как причины выявленных изменений. Мы полагаем, что последние у всех наших больных, скорее всего, обусловлены ТМА миокарда. Верификация микроангиопатического поражения миокарда всегда затруднительна. Однако, принимая во внимание развитие сердечно-сосудистой патологии во время острого эпизода классической ТМА, характерную гистологическую картину почечной ТМА, выявленную при биопсии почек у больных 2, 3 и 4, отсутствие поражения крупных коронарных артерий по данным КАГ у больного 1 с несомненным инфарктом миокарда, наше предположение о наличии ТМА миокарда у всех пациентов с аГУС представляется абсолютно оправданным.
Всем представленным в настоящей работе пациентам во время острого эпизода аГУС проведена плазмотерапия, до настоящего времени остающаяся средством первой линии в лечении практически любой ТМА, в первую очередь ТТП и аГУС [24]. Несмотря на то что эффективность лечения СЗП при аГУС не превышает 50%, мы полагаем, что своевременно начатая плазмотерапия в режимах плазмообмена или инфузии (что в свою очередь связано с быстрой диагностикой ТМА), особенно в сочетании с введением гепаринов, улучшает прогноз. В пользу этого свидетельствует достижение ремиссии аГУС у больных 1 и 5 с полным восстановлением функции всех пораженных органов. У пациентов же 2, 3 и 4, которым диагноз был установлен с опозданием и объем плазмотерапии у которых был невелик, развилась почечная недостаточность, в т.ч. в двух случаях – терминальная, требующая диализной терапии, хотя все экстраренальные проявления, включая ТМА миокарда, были купированы.
Заключение
Представленные наблюдения свидетельствуют о возможности поражения сердца у больных аГУС. Как следует из нашего опыта, микроангиопатическое поражение миокарда, по-видимому, следует считать одним из наиболее частых экстраренальных проявлений аГУС, хотя до настоящего времени оно практически не изучено. Распространенность поражения сердца и степень его влияния на общий прогноз у больных аГУС до сих пор не известны, несмотря на имеющиеся основания считать микроангиопатическое поражение миокарда прогностически неблагоприятным фактором при ТМА [9, 25]. В связи с отсутствием характерной клинической картины общепринятых критериев обследования данных пациентов далеко не всегда удается выявлять признаки, свидетельствующие о нарушении сердечной функции. Однако нам представляется, что всем больным с несомненными лабораторными признаками микроангиопатического гемолиза и тромбоцитопенией целесообразно включать в план обследования определение тропонина I.
Важность этого показателя обусловлена тем, что его уровень, превышающий 0,2 нг/мл в момент острого эпизода ТМА даже у пациентов без явных признаков поражения сердца, следует рассматривать как фактор риска развития острого инфаркта миокарда у этого контингента больных, причем риск увеличивает сочетание повышенного уровня тропонина с уровнем ЛДГ, превышающим 1000 ЕД/л [9]. Другим важным диагностическим методом, безусловно, является Эхо-КГ, которую мы также рекомендуем использовать всем больным, поступающим в стационар с подозрением на ТМА, для исключения бессимптомно протекающего поражения сердца.


