
Введение
Атипичный гемолитико-уремический синдром (аГУС), не связанный с кишечной инфекцией, занимает около 10 % в общей структуре гемолитико-уремического синдрома и представляет собой редкое заболевание, проявляющееся микроангиопатической гемолитической анемией (МАГА), тромбоцитопенией и развитием почечной недостаточности. В последние годы было установлено, что причиной развития аГУС является наследственно обусловленный дефект регуляторных протеинов альтернативного пути активации комплемента (фактора Н, мембранного кофакторного протеина — МСР, фактора I, фактора В) либо, реже, присутствие антител к фактору Н, что приводит к хронической неконтролируемой активации комплемента и развитию комплемент-зависимой тромботической микроангиопатии (ТМА) [1—3].
Сегодня аГУС рассматривают как катастрофическое угрожающее жизни заболевание генетической природы, которое характеризуется частыми рецидивами и неблагоприятным прогнозом: 75 % больных либо умирают в момент острого эпизода, либо демонстрируют быстрое развитие почечной недостаточности, достигающей степени терминальной ХПН в течение года от начала болезни [3, 4]. Менее 20 % случаев аГУС являются семейными, т. е. развивающимися по крайней мере у двух членов одной семьи [2]. АГУС, диагностированный у пациентов, не имеющих семейного анамнеза, классифицируют как спорадический. Последний, как правило, развивается после воздействия таких “пусковых” факторов, как инфекции, злокачественные новообразования, некоторые лекарственные средства (оральные контрацептивы, антибиотики, ингибиторы кальцийнейрина, тиклопидины и др.) [5]. Развитие аГУС возможно при ряде системных болезней (антифосфолипидный синдром, системная красная волчанка, системная склеродермия).
В 12—31 % случаев аГУС связан с беременностью или родами. Установлено, что аГУС, ассоциированный с беременностью (Б-аГУС), чаще всего — в 74% случаев — развивается в III триместре и раннем послеродовом периоде, тогда как на I триместр приходится до 11 % случаев, на II — до 15 % Б-аГУС [6]. По современным представлениям, для диагностики аГУС, как и других ТМА, достаточно только тромбоцитопении и МАГА. В связи с этим Б-аГУС часто бывает сложно отличить от преэклампсии и особенно HELLP-синдрома, для которых также характерно появление тромбоцитопении и микроангиопатического гемолиза, что обусловливает крайнюю редкость постановки диагноза аГУС во время беременности. Приводим собственное наблюдение атипичного ГУС, развившегося у беременной женщины.
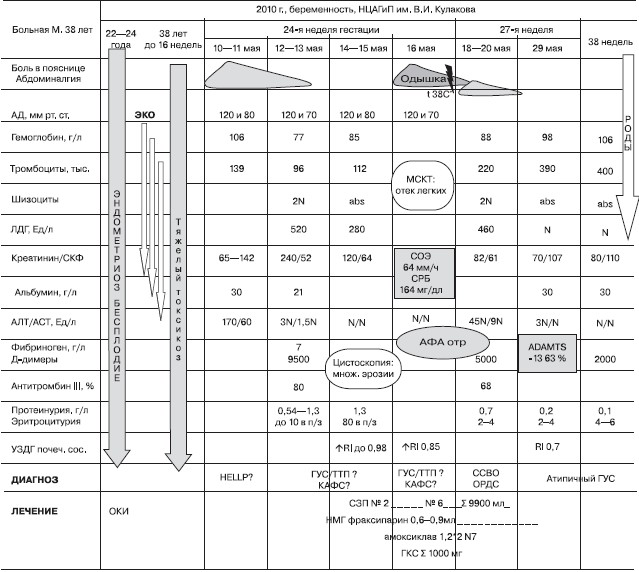
Пациентка М. 38 лет(см. рисунок). Более 20 лет страдала рецидивирующей инфекцией мочевыводящих путей (ИМП) с редкими обострениями. В возрасте 22 лет диагностирован эндометриоз, по поводу которого в течение трех лет принимала оральные контрацептивы, входившие в состав комплексной терапии. В связи с первичным бесплодием дважды предпринимала попытки экстракорпорального оплодотворения (ЭКО), оказывавшиеся неудачными. Данная (первая) беременность наступила после третьей попытки ЭКО. Первый триместр осложнился развитием тяжелого токсикоза с рвотой до 8—10 раз в сутки. Явления токсикоза окончательно исчезли по достижении 16 недель гестации. На сроке 24 недель после погрешности в диете развился умеренно выраженный болевой синдром в поясничной области — преимущественно справа, в связи с чем 11.05.2011 госпитализирована в роддом по месту жительства. Диагноз ИМП был отвергнут, болевой синдром купирован внутримышечным введением спазмолитиков. При обследовании в тот же день впервые выявлены повышение уровня сывороточного креатинина (Скр) до 142 мкмоль/л (по данным обменной карты, за неделю до обращения Скр составлял 65 мкмоль/л), признаки цитолиза с преобладанием АЛТ (АЛТ 170 ЕД/л, АСТ 60 ЕД/л) в отсутствие явлений печеночноклеточной недостаточности и холестаза, анемия (гемоглобин [Нb] 106 г/л) и тромбоцитопения (139 тыс. в 1 мкл). Суточный диурез не уменьшался, изменений в анализе мочи не было. При ультразвуковом исследовании почек данных за мочекаменную болезнь не получено. В связи с предположительным диагнозом HELLP — синдрома 12. 05. пациентка переведена в Научный центр акушерства, гинекологии и перинатологии им. В.И. Кулакова (НЦАГиП). При поступлении отмечены нарастание Скр до 240 мкмоль/л в отсутствие олигурии, резкое снижение Нb до 77 г/л и числа тромбоцитов до 96 тыс. в 1 мкл, сохраняющиеся признаки умеренного цитолиза. Выполненные для уточнения характера анемии прямая и непрямая пробы Кумбса были отрицательными, отмечено повышение уровня лактатдегидро- геназы (ЛДГ) до 520 ЕД/л (норма — 207—414), отсутствие свободного гемоглобина в крови. В мазке периферической крови обнаружены шизоциты в количестве 0,1 % (норма — до 0,05 %). Впервые выявлен умеренный мочевой синдром: протеинурия 0,54 г/л, эритроцитурия до 10 в п/з. Артериальной гипертензии, а также признаков страдания плода не зарегистрировано. Обсуждался диагноз RELLP-синдрома, однако отсутствие нарушений фето- и маточно-плацентарного кровотока заставило усомниться в нем, в связи с чем от досрочного родоразрешения было решено воздержаться до получения результатов полного обследования. К вечеру 12.05. пациентка переведена в отделение реанимации и интенсивной терапии (ОРиТ), где постоянно мониторировали АД, биохимические показатели крови, гемоглобин, тромбоциты, а также общий анализ мочи. 13.05 уровень азотемии оставался стабильным (Скр — 220 мкмоль/л), скорость клубочковой фильтрации (СКФ) по данным пробы Реберга была снижена и составила 52 мл/мин, отмечено быстрое нарастание протеинурии до 1,3 г/л, снижение уровня общего белка до 51 г/л, альбумина — до 21 г/л. Была также отмечена активация внутрисосудистого свертывания крови, не свойственная данному сроку гестации: фибриноген 7 г/л (N 3,5—6,9), Д-димер 9500 мг/дл (N до 1200), уровень антитромбина III (АТ III) 80% (N 80—120). Тяжелая Кумбс-негативная анемия с шизоцитозом и высоким уровнем ЛДГ свидетельствовала о микроангиопатическом гемолизе, что в сочетании с тромбоцитопенией и острым почечным повреждением позволило диагностировать ГУС/ТТП. Для уточнения диагноза исследована кровь на ADAMTS-13, уровень которого составил 63 % (референсные значения — 90—113 %), что позволило исключить ТТП. Отрицательные серологические маркеры АФС (“волчаночный” антикоагулянт, антитела к кардиолипину и бета-2 гликопротеину-1) послужили основанием к исключению катастрофического АФС.
Рисунок. График анамнеза.
Утром 14.05 у больной развилась безболевая макрогематурия, для выяснения причины которой была выполнена цистоскопия, выявившая множественные мелкие эрозии слизистой мочевого пузыря, что не противоречило диагнозу ТМА и трактовалось как ишемическое повреждение слизистой мочевого пузыря вследствие генерализации микроангиопатического процесса.
В связи с невозможностью проведения нефробиопсии для верификации диагноза ренальной ТМА проведена УЗДГ почечных артерий, имеющая высокую информативность в неинвазивной диагностике ГУС благодаря наличию специфических для микроангиопатического поражения почек допплерографических признаков [7]. Эти признаки в виде обеднения коркового кровотока в режиме цветового допплеровского картирования (ЦДК) и резкого повышения индексов резистивности (RI) (до 0,9—0,98 при N до 0,7) позволили подтвердить диагноз.
Принимая во внимание сочетание МАГА, умеренной тромбо-цитопении, почечной недостаточности и клинических признаков поражения других органов (печень, мочевой пузырь) в отсутствие поражения ЦНС 14.05, через двое суток от момента поступления больной в Центр, был установлен диагноз атипичного ГУС. В тот же день проведен первый сеанс плазмообмена с объемом эксфузии 900 мл и восполнением адекватной дозой свежезамороженной плазмы (СЗП). Одновременно начата антикоагулянтная терапия низкомолекулярным гепарином (НМГ) — фраксипарином 0,6 мл/сут. После первого сеанса плазмозамещающей терапии к вечеру 14. 05. уровень Скр снизился до 160 мкмоль/л, уровень Hb повысился до 85 г/л, число тромбоцитов — до 112 тыс. в 1 мкл, нормализовалось значение ЛДГ (280 ЕД/л). Повторная УЗДГ почечных сосудов в режиме ЦДК продемонстрировала улучшение коркового кровотока (значения RI оставались высокими, но снизились до 0,8). После повторного введения 15.05. СЗП в объеме 600 мл уровень Скр достиг 120 мкмоль/л, СКФ увеличилась до 64 мл/ мин. В связи с отчетливой положительной динамикой со стороны почечного процесса было решено сделать перерыв в лечение СЗП из-за опасности развития синдрома Trali (острое повреждение легких, нередко диагностируемое у беременных как осложнение введения СЗП). В то же время продолжалась антикоагулянтная терапия, причем с учетом сохраняющейся активацией внутрисосудистого свертывания доза фраксипарина была увеличена до 0,9 мл/сут.
Однако, несмотря на проведенное лечение НМГ, 16.05. появились выраженный нейтрофильный лейкоцитоз со сдвигом до юных форм, достигающий 17 тыс., увеличение СОЭ до 64 мм/ч, уровень СРБ составил 102 мг/л (N до 0,5). Присоединение к терапии антибиотиков широкого спектра действия (амоксиклав 1,2 г в/м дважды в сутки) не повлияло на течение болезни. Состояние больной ухудшалось: появились лихорадка до 38,2 °С, одышка (ЧДД до 22 в минуту), крепитация в нижних отделах легких; нарастали воспалительные показатели, уровень СРБ достиг 164 мг/л. Заподозрено развитие пневмонии, выполнена КТ легких, выявившая признаки умеренно выраженного альвеолярного отека. Диагностирован острый респираторный дистресс-синдром (ОРДС) взрослых в рамках синдрома системного воспалительного ответа (ССВО), в связи с чем было решено начать внутривенное введение метипреда по 250 мг/сут. В течение трех дней от начала такой терапии лабораторные показатели воспалительного ответа уменьшились (уровень СРБ снизился до 45 мг/л, лейкоцитоз — до 12 тыс.), одышка и аускультативные феномены отека легких исчезли. Проводился динамический контроль за лабораторными показателями: протеинурия уменьшилась до 0,7 г/л, гематурии не наблюдалось, уровень Скр продолжал снижаться и к 18.05 достиг нормальных значений (82 мкмоль/л), однако СКФ оставалась сниженной (61 мл/мин). 17.05 зафиксированы физиологические значения трансаминаз (АЛТ 35 Ед/л, АСТ 17 ЕД/л). Количество тромбоцитов нормализовалось (183 тыс. в 1 мкл), но показатели гемоглобина практически не изменились и не превышали 83—88 г/л, персистировали маркеры тромбинемии (Д-димеры до 5000 мг/дл), уровень АТ III снизился до 68 %.
На пятые сутки продолжающейся антикоагулянтной терапии (18.05.) у пациентки появились боли в эпигастральной области, изжога. Вновь отмечено стремительное нарастание выраженности цитолиза (значения АЛТ к 20.05 достигали 620 ЕД/л, АСТ-340 ЕД/л). При этом АД не превышало 120 и 70 мм рт. ст., плод развивался соответственно срокам гестации. Срочно выполненная ЭГДС обнаружила множественные эрозивно-язвенные дефекты слизистой пищевода, желудка и двенадцатиперстной кишки.
Повторное исследование мазка периферической крови демонстрировало присутствие шизоцитов до 0,1 %, вновь отмечено нарастание уровня ЛДГ до 460 ЕД/л. Несмотря на нормализацию числа тромбоцитов и уровня креатинина (202 тыс. в мкл и 74 мкмоль/л соответственно), острое развитие эрозивно-язвенного поражения желудочно-кишечного тракта (ЖКТ), рецидивирующее поражение печени с нарастающим цитолизом при сохраняющейся анемии с шизоцитозом были расценены как генерализация ТМА с вовлечением в микроангиопатический процесс не только печени, но и сосудистого русла ЖКТ. По данным в третий раз выполненной УЗДГ почечных сосудов выявлена псевдонормализация показателей коркового кровотока: на фоне незначительного повышения RI (до 0,7) появились нормо- и низкорезистивные формы спектра, что, с одной стороны, свидетельствовало об активном юкстамедуллярном шунтировании, направленном на компенсацию нарушений локально-почечной гемодинамики, с другой — подтверждало наличие сохраняющейся ишемии паренхимы почек, предполагать которую позволяла низкая СКФ.
Принимая во внимание генерализацию микроангиопатического тромбообразования с вовлечением сосудистого русла ЖКТ, развитие синдрома системного воспалительного ответа после четырех дней перерыва с 20.05 была возобновлена плазмотерапия: проведен сеанс плазмообмена с объемом удаления и замещения СЗП из расчета 30 мл/кг/сут. Повторно проведен “пульс” метипредом в дозе 500 мг. В течение трех последующих дней продолжались процедуры плазмообмена с ежедневной заменой 2600 мл плазмы, в результате чего купированы проявления гемолиза (отсутствие шизоцитов, нормализация ЛДГ), наметилась тенденция к нормализации гемоглобина (96 г/л), уровень АЛТ снизился до 130 ЕД/л, уровень АСТ нормализовался. Затем в течение следующих 5 дней выполнялись сеансы плазмафереза с удалением по 600 мл плазмы и возмещением коллоидными растворами. Показатели почечной функции стойко нормализовались, СКФ практически достигла нормальных для беременности значений (Скр —70 мкмоль/л, СКФ — 107 мл/мин), протеинурия снизилась до 0,2 г/л. При контрольной УЗДГ почечных сосудов отмечена нормализация показателей внутрипочечной гемодинамики, изменений коркового кровотока не зарегистрировано. Число тромбоцитов составило 390 тыс. в 1 мкл, уровень гемоглобина — 98 г/л. Развитие плода соответствовало срокам беременности. Через 17 дней с момента госпитализации, 29 мая, на сроке беременности 27 недель пациентка была выписана на амбулаторное лечение по месту жительства с нормальными показателями функции почек и картины крови и без признаков поражения печени, ЖКТ, мочевого пузыря, легких. На основании характера течения заболевания, его клинико-лабораторных особенностей, ответа на проводимую терапию был установлен диагноз "атипичный гемолитико-уремический синдром: микроангиопатическая гемолитическая анемия, тромбоцитопения, острое почечное повреждение, поражение легких, печени, желудочно-кишечного и мочевого трактов. Беременность — 27 недель (ЭКО)" . Рекомендовано продолжить антикоагулятную терапию (фраксипарин 0,9 мл/сут) под контролем клинико-лабораторных показателей, что и выполняла в течение двух с половиной месяцев.
На сроке 37 недель пациентка вновь обратилась в НЦАГиП для планового родоразрешения. При контрольном обследовании признаков гемолиза и тромбоцитопении не зарегистрировано (Hb 110 г/л, тромбоциты 360 тыс. в мкл, ЛДГ 315 ЕД/л), изменений в анализе мочи не выявлено, функция почек оставалась нормальной (Скр 85 мкмоль/л, СКФ 125 мл/мин), цитолиз отсутствовал (АЛТ 15 ЕД/л, АСТ 22 ЕД/л). Роды проведены через естественные родовые пути (излитие вод на сроке 38 недель), родилась живая доношенная девочка весом 3450 г, ростом 52 см с оценкой по шкале Апгар 8/9 баллов. В послеродовом периоде возобновлена антикоагулянтная терапия. Была выписана на 7-е сутки с рекомендацией продолжить антикоагулянтную терапию в течение 6 недель после родоразрешения.
Известно, что в течение последнего года пациентка чувствует себя хорошо, гемодинамические и лабораторные показатели стабильно нормальные. Ребенок развивается соответственно возрасту, находится на грудном вскармливании.
Обсуждение
Продемонстрированное нами наблюдение иллюстрирует трудности диагностики, особенности клинического проявления и течения “акушерского” аГУС, а также возможности благоприятного исхода этого тяжелого жизнеугрожающего заболевания для матери и плода при условии своевременно установленного диагноза и эффективной плазмотерапии.
Полиморфная клиническая картина болезни, представленная признаками поражения почек, печени, мочевого пузыря, желудочно-кишечного тракта, а затем и легких, требовала проведения для пациентки с МАГА и тромбоцитопенией дифференциального диагноза между различными микроангиопатическими синдромами, в т. ч. аГУС, тромботической тромбоцитопенической пурпурой (ТТП), катастрофическим АФС (КАФС), острым ДВС-синдромом, но в первую очередь HELLP-синдромом. Исключение HELLP-синдрома представлялось особенно важным, поскольку этот диагноз требует немедленного родоразрешения, промедление с которым может иметь катастрофические последствия для жизни женщины. Сочетание гипертрансаминаземии, отражающей повреждение печени, микроангиопатического гемолиза, тромбоцитопении, хотя и умеренно выраженной, позволяло в данном случае обсуждать диагноз HELLP-синдрома, несмотря на относительно небольшой для его манифестации срок беременности (24 недели) и отсутствие ключевого симптома преэклампсии — артериальной гипертонии, поскольку, по данным Katz и соавт. (2009), HELLP-синдром и эклампсия могут развиваться в отсутствие типичных признаков тяжелой преэклампсии и быть первым проявлением генерализованной жизнеугрожающей ТМА [8]. Известно, что развитие HELLP- синдрома характерно для более поздних сроков беременности — в среднем 37—38 недель — или для раннего послеродового периода. Однако основным фактором, позволившим исключить этот диагноз, оказался не срок гестации, а отсутствие признаков страдания плода и нормальные показатели фето- и маточно-плацентарного кровотока, чего невозможно ожидать от пациентки с HELLP-синдромом с учетом ведущей роли плаценты в развитии этой патологии. Именно понимание важности ее удаления для спасения жизни женщины диктует необходимость “агрессивной” тактики ведения пациенток с HELLP-синдромом (незамедлительное родоразрешение путем кесарева сечения независимо от сроков беременности) и делает неоправданным консервативный подход к их лечению. Установлено, что при HELLP-синдроме применение плазмозамещающей терапии требуется довольно редко — лишь в случаях отсутствия улучшения состояния пациенток в течение 72 часов после кесарева сечения, поскольку поражение сосудов микроциркуляторного русла печени в большинстве случаев довольно быстро регрессирует самостоятельно после экстренного родоразрешения [6]. Напротив, даже при подозрении на Б-аГУС требуется незамедлительное начало обменного переливания плазмы, а родоразрешение не только не способствует регрессу симптоматики, но и в ряде случаев усугубляет клиническую картину болезни, поскольку оперативное вмешательство может стать дополнительным триггером активации комплемента. Несвоевременное начало плазмотерапии или отказ от ее применения может стать причиной летального исхода: до внедрения в практику плазмообмена материнская смертность при аГУС достигала 95 % [9]. Именно поэтому разграничение Б-аГУС и HELLP-синдрома является первоочередной задачей для беременных с признаками ТМА.
Другой патологией, симптомы которой напоминали клинические проявления болезни в данном случае, несмотря на отсутствие признаков поражения ЦНС, является ТТП. Ее развитие у беременных, как и в популяции в целом, обусловлено приобретенным дефицитом металлопротеазы ADAMTS-13, расщепляющей сверхкрупные мультимеры фактора фон Виллебранда (фВ) и являющейся, таким образом, важным фактором ограничения тромбообразования в сосудах микроциркуляторного русла различных органов. Однако из-за специфических гестационных изменений в системе “ фВ и ADAMTS-13” особенности патогенеза ТТП, развивающейся во время беременности, изучены недостаточно [10]. Установлено, что беременности свойственно нарушение баланса между уровнем фВ и ADAMTS-13: активность фактора Виллебранда на протяжении всего гестационного процесса неуклонно нарастает, достигая максимума (до 200—500 % нормы) преимущественно в конце второго-третьем триместрах в основном за счет формирования сверхкрупных мультимеров, обладающих повышенной тромбогенностью. С другой стороны, показатели плазменной активности ADAMTS-13 начиная с конца первого триместра постепенно снижаются и в раннем послеродовом периоде имеют наиболее низкие значения [11,12]. По-видимому, дисбаланс между фВ и ADAMTS-13 во время беременности наряду с другими изменениями в системе гемокоагуляции можно рассматривать как адаптационную реакцию, направленную на минимизацию кровопотери в родах. Можно предположить, что избыточное нарастание уровня фВ при неадекватно низком уровне ADAMTS-13, недостаточном для полноценного противостояния ингибиторному эффекту анти-ADAMTS-13-антител, обусловливает наиболее высокий риск развития ТМА именно в конце второго — начале третьего триместров [12]. В крупном исследовании J.N. Martin и соавт, включившем 166 случаев ТТП, ассоциированной с беременностью, как и в представленном наблюдении, ТМА развилась преимущественно в те же сроки — в среднем на 23-й неделе гестации [13], что давало основания для исключения ТТП у нашей пациентки. Однако небольшое снижение уровня ADAMТS-13 (63 %), соответствующее гестационной норме для данного срока [12], а также отсутствие неврологической симптоматики позволили отвергнуть этот диагноз.
Преимущественное поражение сосудов малого калибра, в короткие сроки (от нескольких часов до нескольких дней) приводящее к развитию полиорганной недостаточности, характерно для КАФС [14]. Предполагать этот диагноз в данном случае можно было на основании полиморфной клинической картины, представленной симптомами поражения почек, печени, легких, желудочно-кишечного тракта и мочевого пузыря. Необычная локализация микроциркуляторного поражения, отмеченная в данном случае, является отличительной особенностью КАФС. Дополнительным аргументом в пользу возможности этого диагноза были эндометриоз, первичное бесплодие и неудачи двух первых попыток ЭКО, нередко ассоциированные с присутствием антифосфолипидных антител [15]. Однако проведенное обследование не выявило серологических маркеров АФС, что позволило исключить диагноз КАФС.
Наконец, клинико-лабораторные проявления заболевания, в т. ч. избыточно высокие показатели Д-димера, не соответствующие сроку гестации, требовали проведения дифференциальной диагностики с декомпенсированным ДВС-синдромом. Однако при беременности развитие ДВС-синдрома осложняет различные виды акушерской патологии (преэклампсия, антенатальная гибель плода, преждевременная отслойка плаценты и др.), отсутствующие у представленной пациентки, что делало этот диагноз маловероятным. Кроме того, МАГА довольно редко развивается у больных с ДВС-синдромом (около 15 % случаев), причем выраженная анемия с шизоцитозом не бывает ранним его проявлением, как это имело место в настоящем наблюдении. Безусловно признаки активации внутрисосудистого свертывания крови, имеющиеся у пациентки, не позволяют исключать ДВС-синдром, осложнивший аГУС.
Таким образом, проведенное в течение первых 2 суток пребывания пациентки в НЦАГиП им. В.И. Кулакова обследование позволило отвергнуть несколько различных вариантов ТМА (HELLP-синдром, ТТП, КАФС, острый ДВС-синдром), развитие которых возможно при беременности, и установить диагноз атипичного ГУС. Особенностями заболевания в данном случае были его развитие во втором триместре, спектр клинических проявлений, быстрый ответ на плазмотерапию и благоприятный исход беременности для матери и ребенка.
Исследованиями последних лет установлено, что ассоциированный с беременностью аГУС составляет около 20 % в структуре аГУС, причем 75 % случаев акушерского аГУС приходится на послеродовый период [6,11]. Развивающийся в это время аГУС характеризуется агрессивным течением и неблагоприятным прогнозом. Так, из 21 пациентки с Б-аГУС, наблюдаемых F. Fakhouri и соавт., только у четверых заболевание манифестировало в 1—2-м триместрах, у остальных — в сроки от 3 дней до 3 месяцев после родов; лишь у 1 пациентки была достигнута полная ремиссия болезни, у 15 женщин — в исходе острого эпизода (у большинства в течение месяца) развилась терминальная ХПН (тХПН), еще у пятерых сохранялись признаки ХБП, прогрессирование которой в 3 случаях привело к развитию тХПН в течение 19—36 месяцев [11]. Преимущественное развитие аГУС в послеродовом периоде на первый взгляд кажется парадоксальным, поскольку известно, что беременность per seслужит мощным триггером активации комплемента. При этом плацента является потенциальной мишенью комплемент-опосредованной иммунной атаки, что создает угрозу гибели плода. Последнее положение подтверждается обнаружением компонентов комплемента (С3b, С4b) в плаценте как при нормальной, так и при патологической беременностях [16]. Риск комплемент-зависимого повреждения плаценты, как оказалось, уравновешивает существование локального защитного механизма, блокирующего нежелательный эффект активации комплемента. При нормальной беременности неконтролируемую активацию последнего предотвращают три основных регуляторных белка: DAF (decay-accelerated factor), играющий ведущую роль в “противо-комплементарной” защите плаценты, MCP (membrane cofactor protein) и CD59. Все они экспрессированы на поверхности трофобласта и ингибируют комплемент, снижая активность С3 конвертазы альтернативного пути [10]. У пациенток с нарушенной регуляцией альтернативного пути вследствие мутаций генов CHF, CFI,C3 и MCP во время беременности эффективный контроль активации комплемента и локальную защиту плаценты обеспечивает DAF [11]. Напротив, после родов имеющееся воспаление, обусловленное наличием обширной раневой поверхности, попадание в материнский кровоток клеток плода, кровотечение, инфекция ведут к системной активации альтернативного пути, дополнительный вклад в которую вносит элиминация эффективных регуляторных механизмов, которые обеспечивала плацента per se. Совокупность этих факторов индуцирует развитие аГУС у лиц с генетической предрасположенностью [11].
“Раннее” (во 2-м триместре) развитие ассоциированного с беременностью аГУС у представленной пациентки, таким образом, скорее является исключением, чем правилом. Мы не смогли выполнить больной генетические исследования, однако даже при наличии такой возможности у 30—50 % пациентов не удается идентифицировать мутации в генах регуляторных белков комплемента, ведущие к развитию аГУС [2], и для установки диагноза, таким образом, достаточно лишь характерной клинико-лабораторной картины, как это имело место у нашей пациентки. Причина “раннего” развития аГУС в представленном наблюдении осталась неизвестной. Можно предполагать, что триггером послужила беременность как таковая, однако локальные механизмы защиты плаценты от неконтролируемой активации комплемента оказались неповрежденными, в пользу чего свидетельствуют нормальные показатели маточно- и фето-плацентарного кровотока даже в критической для матери ситуации, а также исход беременности, завершившейся рождением здорового доношенного ребенка. Достижение ремиссии аГУС при развитии тяжелой полиорганной патологии в данном случае, по-видимому, можно объяснить сочетанием плазмотерапии с эффективной “антикомплементарной” функцией локальных регуляторных протеинов, фиксированных на поверхности трофобласта.
Преимущественное поражение почек в дебюте заболевания в сочетании с признаками МАГА и тромбоцитопении позволило нам диагностировать аГУС и начать лечение свежезамороженной плазмой сразу после установки диагноза, 14.05, 2011, т. е. на вторые сутки от момента поступления в клинику. Быстрый положительный эффект, выраженный не только в улучшении функции почек, но и в повышении уровня гемоглобина и числа тромбоцитов уже после первой процедуры плазмаобмена, создал в данном случае обманчивое впечатление благополучия, что привело к ошибочному решению прервать лечение СЗП после второго сеанса, не дождавшись полного прекращения микроангиопатического гемолиза и стабилизации количества тромбоцитов. В соответствии с имеющимися сегодня рекомендациями по лечению аГУС [17] прекращение плазмотерапии возможно лишь в том случае, если уровень ЛДГ, достигший нормальных значений, не нарастает, а количество тромбоцитов не снижается после нормализации в течение 2 дней подряд. Несоблюдение этого правила может приводить к ухудшению состояния пациента, как это и произошло в данном случае. Дальнейшее течение заболевания с вовлечением в патологический процесс других органов, свидетельствующим о генерализации ТМА, развитие синдрома системного воспалительного ответа с ОРДС взрослых, быстрый рецидив гемолиза, несмотря на нормализацию уровня креатинина крови и числа тромбоцитов, подтвердили ошибочность решения о перерыве в лечении СЗП, продемонстрировав неэффективность применения НМГ в терапевтических дозах в отсутствие плазмотерапи. Ее возобновление после четырехдневного перерыва вновь сопровождалось быстрым положительным эффектом и привело к достижению полной ремиссии заболевания уже через неделю от начала повторного введения СЗП. Характер течения болезни, особенности ответа на терапию и исход аГУС у нашей пациентки подтверждают данные P. Ruggenenti и соавт., свидетельствующие о том, что исход поражения почек зависит не от конкретного вида ТМА, а определяется прежде всего ранним началом адекватной плазмозамещающей терапии [18]. Наряду с лечением СЗП и внутривенным введением преднизолона, направленным на купирование ССВО, представленная пациентка получала терапию НМГ, которая, с нашей точки зрения, усиливает антикоагулянтный эффект плазмы. Взаимодействие гепаринов с антитромбином III, содержащимся в СЗП, формирует антикоагулянтный комплекс практически немедленного действия, что представляется крайневажным для купирования генерализации тромбообразования в сосудах микроциркуляторного русла. В то же время наряду с антитромботической активностью гепарину свойственны антикомплементарное и противовоспалительное действие [19], что оправдывает его применение при аГУС, поскольку препарат может способствовать уменьшению выраженности ССВО и комплемент-зависимого тромбообразования.
Заключение
Таким образом, представленное наблюдение имеет целью ознакомить практических врачей с особенностями течения, подходами к терапии и исходом атипичного ГУС, развившегося во время беременности. Ассоциированная с беременностью и родами ТМА независимо от срока ее развития ставит перед врачом вопрос о необходимости разграничения различных микроангиопатических синдромов — в первую очередь HELLP-синдрома, но также аГУС, ТТП и катастрофического АФС. Несмотря на сходство клинико-лабораторных проявлений этих видов патологии, подходы к их лечению и тактике ведения беременности различаются, в связи с чем своевременно и четко установленный диагноз необходимо рассматривать как основной фактор, определяющий прогноз и для матери, и для плода.



{kind=link}