
Введение
Гемолитико-уремический синдром (ГУС) по причинам возникновения может быть разделен на инфекционные и неинфекционные формы [1]. Несмотря на то, что наиболее распространена типичная форма ГУС с диарейной продромой, ассоциированная с токсином Шига (STEC), требуется тщательное подтверждение инфекционной этиологии, чтобы вовремя исключить атипичный ГУС (аГУС) [2]. Клинически аГУС характеризуется триадой признаков: Кумбс-негативной гемолитической анемией с наличием фрагментированных эритроцитов (шизоцитов), тромбоцитопенией и острым повреждением почек (ОПП). Указанные признаки являются компонентами тромботической микроангиопатии (ТМА) – распространенной окклюзии сосудов мелкого калибра тромбами, возникшими вследствие повреждения эндотелия [3].
аГУС чаще всего имеет в основе генные мутации, приводящие к дисфункции каскада комплемента с неконтролируемой активацией альтернативного пути [4]. При активации комплемента образуется конвертаза C3bBb, которая приводит к превращению C3 в C3b и нарушается процесс подавления избыточной активности системы комплемента, что приводит к реализации повреждающего действия конечных продуктов его альтернативного пути на клетки эндотелия с развитием ТМА [5]. Гистологическими находками при ТМА являются утолщение стенок капилляров и артериол с отеком и десквамацией эндотелия, субэндотелиальное отложение белка и клеточного детрита, тромботическая обтурация просвета капилляров [6].
Плазмотерапия эффективна в наибольшей степени при мутациях большого количества функциональных белков [6, 7]. Предпочтителен мембранный плазмаферез с объемом замещения 50–60 мл на кг массы тела, альтернативный вариант – инфузия свежезамороженной плазмы (СЗП) в объеме 10–20 мл/кг. В большинстве случаев функция почек полностью восстанавливается. Промежуток времени между рецидивами колеблется иногда от нескольких недель до многих лет [7].
Клинический случай
Пациент Д. 19 лет, 21.12.2023 доставлен в приемное отделение терапии с жалобами на выраженную слабость, недомогание, тошноту, рвоту с прожилками крови, носовые кровотечения, боль в животе и в горле, плохой аппетит, «эпизоды покраснения мочи» и снижение ее количества, повышение температуры до 38◦С. Пациент в течение недели лечился амбулаторно по поводу острой респираторной вирусной инфекции (ОРВИ), отмечал лихорадку, боль в горле, ломоту во всем теле, принимал жаропонижающие и нестероидные противовоспалительные средства.
При осмотре исключена урологическая патология (при обследовании, по данным ультразвукового исследования почек: подковообразная почка и спленомегалия, выявлено повышение уровней креатинина до 242,7 мкмоль/л, мочевины до 15,87 ммоль/л. Пациент госпитализирован в Центр по лечению пациентов с острой почечной недостаточностью ГБУЗ НСО «ГКБ № 34», Новосибирск.
При поступлении в отделение: состояние пациента тяжелое. Тяжесть состояния обусловлена интоксикационным, абдоминальным синдромами, развитием ОПП. Параклинически при поступлении: в общем анализе мочи – цвет красный, протеинурия до 0,5 г/л, лейкоцитурия до 13 в п/зр, гематурия; в общем анализе крови выявлена тромбоцитопения до 14×109, в биохимическом анализе крови – умеренный синдром цитолиза, гиперазотемия (см. таблицу).
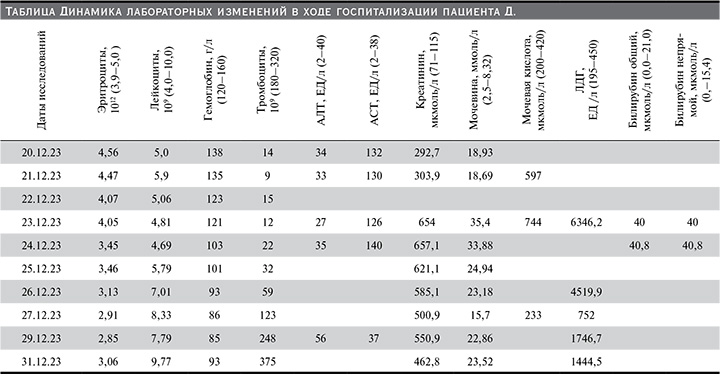
По данным фиброгастродуоденоскопии – эрозивный гастрит. Весь период стационарного лечения у пациента, по данным гемостазиограммы, наблюдалась умеренная гипокоагуляция: активированное частичное тромбопластиновое время не выше 40,5 секунд (референсные значения – 26–36 секунд), наблюдалось прогрессирование почечной дисфунции (см. таблицу), что потребовало проведения сеансов заместительной почечной терапии (ЗПТ). Всего проведено четыре сеанса ЗПТ методом острого гемодиализа (23.12.2023, 24.12.2023, 25.12.2023, 28.12.2023). Плазмотрансфузии проводили 23.12.2023, 25.12.2023, 27.12.2023 в объеме 20 мл на кг массы тела. На фоне проведения плазмотранфузии отмечался быстрый рост уровня тромбоцитов в крови (см. таблицу), переливания тромбоцитарной массы не потребовалось (трансфузии тромбоконцентрата у пациентов с аГУС могут провоцировать новые эпизоды микротромбообразования). Был запланирован сеанс плазмообмена (MPS+PE мембранная плазмосепарация с плазмообменом, аппарат Mulifiltrate, Fresenius), но ввиду стойкого положительного эффекта от плазмотрансфузий проведения процедуры не потребовалось. Выявлено снижение активности ADAMTS-13 до 48% (норма – 80–110%), что характерно для аГУС.
С 21.12.2023 по 27.12.2023 у пациента Д. отмечалась олигурия, восстановление диуреза и нормализация температуры произошли сразу же после первой плазмотрансфузии. Пациент Д. был выписан 11.01.2024 (на 22-е сутки) на амбулаторный этап в удовлетворительном состоянии с полным восстановлением диуреза и почечной функции.
Верифицирован заключительный клинический диагноз: «ГУС, атипичная форма, тяжелое течение. ОПП, стадия 3». Сопутствующий: «аномалия развития мочевыводящей системы: подковообразная почка. Эрозивный гастрит».
Обсуждение
Клиническая картина аГУС характеризуется полиморфизмом симптомов и развитием ТМА. У пациента Д. развилась ТМА с тромбоцитопенией до 9х109, гемолитической анемией (снижение гемоглобина до 85 г/л и появление шизоцитов в мазке периферической крови до 2%), ОПП (олигоанурия и гиперазотемия). У пациента Д. значение ADAMTS-13 составило 48% (при аГУС уровень маркера более 10%, но менее 50%). Для пациентов с аГУС характерны мутации в генах регуляторных белков и факторов комплемента. Выявление данных генетических изменений наиболее информативно для определения прогноза трансплантации почек, поэтому в представленном клиническом случае исследования полиморфизмов генов не проводилось. Эффективной терапией при аГУС считается введение свежезамороженной плазмы, на фоне плазмотерапии отмечался регресс в течении ОПП, тромбоцитопении и анемии, нормализация уровня лактатдегидрогеназы. Пациенту на 3-и сутки госпитализации начали сеансы ЗПТ в режиме вено-венозной гемофильтрации с ограничением гепарина ввиду рвоты с прожилками крови и носовыми кровотечениями. В рассмотренном случае терапия экулизумабом не была начата в связи с быстрым восстановлением почечной функции. Ретроспективно можно констатировать, что позднее начало комплементблокирующей терапии не оказало ожидаемого положительного влияния на течение заболевания. У пациента Д. были признаки вялотекущей инфекции, что также является ограничением для применения экулизумаба.
Заключение
Представленный случай демонстрирует сложность дифференциальной диагностики аГУС на ранних этапах заболевания на фоне триггера ОРВИ. Своевременность и быстрота диагностического процесса при аГУС является сложным процессом и может занимать достаточно длительное время.


