
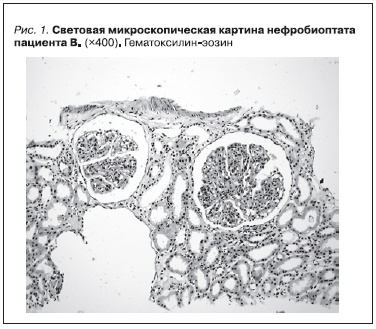
Мембранопролиферативный гломерулонефрит II типа (МПГН-II) характеризуется диффузным утолщением стенок клубочковых капилляров с возможной интерпозицией мезангия в эту область, что приводит к картине двухконтурности (“трамвайный путь”) гломерулярной базальной мембраны
(ГБМ). Имеются выраженная гиперцеллюлярность клубочка и расширение мезангиального матрикса. Особо типичной является электронно-микроскопическая картина, демонстрирующая электронно-плотные интрамембранозные депозиты, состоящие из С3 и продуктов деградации комплемента. По их
наличию данную патологию определяют так же, как и болезнь плотных депозитов (БПД).
Заболевание относится к числу очень редких (2–3 случая на миллион населения) и встречается практически исключительно в возрасте от 5 до 15 лет.
В отличие от мембранопролиферативного гломерулонефрита I типа, считающегося иммунокомплексным заболеванием почек, при МПГН-II доказательств иммунокомплексной природы болезни не имеется. В патогенезе БПД основная роль отводится дисфункции системы комплемента с неконтролируемой активацией его альтернативного пути. У 80 % пациентов с МПГН-II определяется сывороточный С3-нефритический фактор (C3NeF) – аутоантитело к C3bBb, конвертазе альтернативного пути каскада комплемента. Поскольку C3NeF определяется и при МПГН-I более чем у половины больных, основой диагностики является электронная микроскопия биоптата почки.
Когда C3NeF связывается с C3bBb, устойчивость последней к метаболической деградации повышается, активность этой конвертазы сохраняется более длительно, что и поддерживает неконтролируемую активацию альтернативного пути каскада комплемента с низким уровнем С3 и его депозицией в ГБМ.
Помимо этого существует несколько факторов регуляции комплемента, наиболее значимым из которых при описываемой патологии является фактор H (fH) – растворимый гликопротеин, дестабилизирующий конвертазу C3bBb. Активность последней возрастает при дефиците fH, обусловленной мутацией гена на хромосоме 1q32, ответственного за его синтез, или с присутствием антител к fH. Данные нарушения также сопровождаются активацией альтернативного пути комплемента. Наряду с МПГН-II дефицит fH лежит в основе патогенеза некоторых форм атипичного гемолитико-уремического синдрома, часто носящего семейный характер при наличии мутаций fH [1, 2].
Клинически у всех пациентов отмечена гематурия и протеинурия, почти в половине случаев приводящие к нефротическому синдрому. Болезнь носит прогрессирующий характер, и примерно у 70 % больных в течение 10 лет развивается терминальная стадия хронической почечной недостаточности.
К экстраренальным проявлениям МПГН-II относятся изменения на сетчатке, напоминающие старческую дегенерацию с наличием характерных бляшек беловато-желтого цвета (drusen), которые только со временем могут нарушить зрительную функцию.
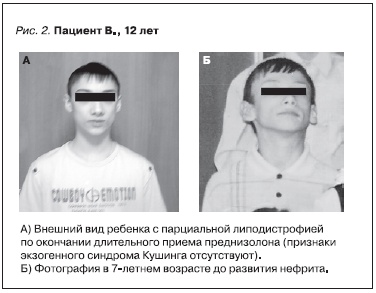
МПГН-II может сопровождаться приобретенной парциальной липодистрофией (ПЛД), преимущественно затрагивающей верхние отделы тела. ПЛД может отмечаться за годы до развития нефрита, и уже в это время наблюдается низкий уровень С3 в сыворотке. Развитие ПЛД связано с отложением активированных продуктов каскада комплемента в жировой ткани и разрушением адипоцитов [3].
Эффективных способов лечения МПГН-II не известно. Контролируемых исследований не проводилось из-за крайней редкости заболевания. Иммуносупрессивная терапия не приводит к увеличению выживаемости больных, хотя в некоторых случаях определенный эффект возможен, вероятно вследствие подавления продукции антител к fH. Умеренной эффективностью обладают сочетанное применение антикоагулянтов и антиагрегантов, а также терапевтический плазмаферез с введением свежезамороженной плазмы. Это может объясняться удалением C3NeF и введением экзогенного fH.
Перспективными новыми методами считаются применение препарата Экулизумаб, моноклонального антитела к протеину C5-комплемента. Экулизумаб ингибирует каскад комплемента за счет снижения активности ряда конвертаз. Ведутся также работы по синтезу рекомбинантного fH.
МПГН-II развивается в почечном трансплантате практически в 100 % случаев, однако болезнь при этом не носит столь тяжелого характера, как в нативной почке, и в отношении трансплантации у таких больных не должно быть полного отрицания [4, 5]. Обсуждается вопрос об эффективности
трансплантации печени при дефиците fH, т. к. последний синтезируется именно в печени.
В отделении нефрологии НЦЗД РАМН в течение 1,5 лет (03.2009–09.2010) с диагнозом “мембранопролиферативный гломерулонефрит II типа (пункционная нефробиопсия 04.2008) с парциальной липодистрофией” наблюдался пациент В. 1996 г. р.
Семейный анамнез у больного отягощен не был. До начала настоящего заболевания мальчик считался практически здоровым.
Дебют заболевания имел место в феврале 2007 г. в возрасте 10 лет с развернутого нефротического синдрома (протеинурия – до 6 г/л, гипопротеинемия – 30–40 г/л, гиперхолестеринемия – более 10ммоль/л, выраженные отеки), гематурии, артериальной гипертензии до 160/100 мм рт. ст. Терапия преднизолоном в дозе 60 мг/сут в течение 2,5 недель, далее в связи с развитием гастродуоденита в дозе 40 мг/сут длительностью ~3 месяцев оказалась неэффективной. При этом развития кушингоидного синдрома не наблюдалось.
В июне 2007 г. по месту жительства проведена нефробиопсия, выявившая мембранопролиферативный гломерулонефрит с выраженными тубулоинтерстициальными изменениями. Был проведен курс лечения циклоспорином А в течение ~1 года с незначительной положительной динамикой. Далее в связи с
отсутствием препарата терапия продолжена приемом мофетила микофенолата в сочетании с преднизолоном 30 мг/48 ч. С целью коррекции артериальной гипертензии мальчик получал ингибитор ангиотензинпревращающего фермента лизиноприл с положительным эффектом.
Спустя ~6 месяцев от начала терапии мофетила микофенолатом отмечено снижение активности заболевания. Протеинурия составила 0,5–1,4 г/сут, уровень общего белка в сыворотке крови – 57 г/л, холестерина – 7,8 ммоль/л. Содержание С3-компонента комплемента в сыворотке крови было снижено до 12,2 мг%. Скорость клубочковой фильтрации (СКФ) соответствовала I стадии хронической болезни почек (ХБП), равной 110,5 мл/мин. Артериальное давление по однократным измерениям не поднималось выше 120/75 мм рт. ст., хотя по данным суточного мониторирования средние показатели в
дневные и ночные часы были повышены (130/92 и 113/77 мм рт. ст. соответственно). Гемоглобин был на уровне 109 г/л. Специальные методы исследования не подтвердили наличия гепатитов и системных заболеваний.
Тогда же были изучены биоптаты почечной ткани, полученные при проведении по месту жительства повторной нефробиопсии в апреле 2008 г. (рис. 1). Был заподозрен МПГН-II. Обращала на себя внимание парциальная липодистрофия лица (рис. 2), подтвержденная рядом фотографий начиная с
возраста 5 лет.
Было продолжено лечение мофетила микофенолатом. Спустя ~1 год от начала указанной терапии вновь отмечено возрастание активности нефротического синдрома. Уровень протеинурии составил 3–6 г/л, общего белка в сыворотке крови – 30–40 г/л, холестерина – более 10 ммоль/л; имели
место выраженные отеки век, голеней, стоп. Была проведена интенсивная симптоматическая и ренопротективная терапия, продолжен прием мофетила микофенолата.
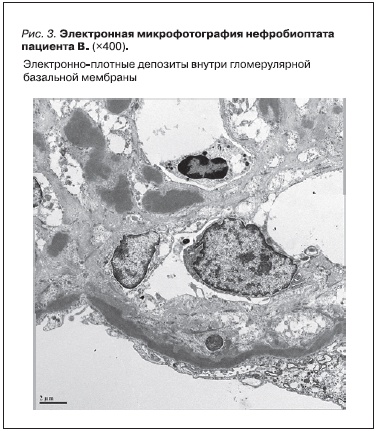
По данным электронной микроскопии был подтвержден МПГН-II с распространенными электроно-плотными депозитами по ходу всей гломерулярной базальной мембраны (рис. 3). Проведен забор крови для молекулярно-генетического исследования, хотя полученные значительно позднее (02.2011)
результаты исследования мутаций в гене fН-комплемента в “горячих точках” их не подтвердили.
Полученные результаты обследования и характер течения заболевания позволили считать лечение иммуносупрессивными препаратами в дальнейшем нецелесообразным. Была рекомендована ренопротективная и симптоматическая терапия, которая затем проводилась нерегулярно.
Спустя 7 месяцев отмечено существенное ухудшение в состоянии ребенка. Имели место выраженные отеки голеней, век. Протеинурия составила 4–6 г/сут, уровень общего белка в сыворотке крови – 35 г/л, альбуминов 12 г/л, холестерина – 17,5 ммоль/л. Содержание С3-компонента комплемента в
сыворотке крови было снижено до 0,21 г/л. СКФ соответ ствовала II стадии ХБП – 78,6 мл/мин. Артериальное давление по однократным измерениям составило 150/90 мм рт. ст., по данным суточного мониторирования средние показатели в дневные и ночные часы были повышены (147/112 и 141/103
мм рт. ст. соответственно). Выявлено расширение полости левого желудочка (50/34 мм), утолщение его стенок (задняя стенка левого желудочка 8,5 мм). Сохранялась анемия: уровень Нв – 108 г/л. Вновь был проведен подбор антигипертензивной терапии. Была использована многокомпонентная схема,
включившая ингибитор ангиотензинпревращающего фермента лизиноприл, антогонист кальция амлодипин, диуретики (гидрохлортиазид, индапамид). Назначены антиагрегантная и антикоагулянтная терапия, препараты кальция, витамина D, железа, эритропоэтина. На этом фоне удалось достичь стабилизации состояния, выраженность отеков существенно снизилась, уровень артериального давления составил 120/90 мм рт. ст., уменьшились размеры левого желудочка (44/26
мм), гемоглобин достиг 123 г/л. Рекомендовано продолжить отмеченную выше терапию, однако в дальнейшем заболевание имело быстропрогрессирующий характер.
Таким образом, особенности течения заболевания (дебют в возрасте 10 лет, высокая активность нефротического синдрома, сохранявшаяся на фоне интенсивной иммуносупрессивной терапии, трудноподдающаяся коррекции артериальная гипертензия, прогрессирующее снижение почечных
функций), низкий уровень С3-компонента комплемента в сыворотке крови, характерные морфологические изменения, наличие парциальной липодистрофии позволили поставить
указанный выше диагноз, несмотря на результаты молекулярно-генетического исследования. Некоторое снижение активности заболевания в начале лечения мофетила микофенолатом, возможно, обусловлено в определенной степени подавлением на фоне указанной терапии С3-нефритического
фактора – антитела, направленного против С3-конвертазы, регулирующей альтернативный путь комплементного каскада, некотролируемая активация которого лежит в основе патогенеза МПГН-II.


