
Тромботическая тромбоцитопеническая пурпура (ТТП) была впервые описана в 1924 г. E. Moschcowitz у 16-летней девушки с лихорадкой, поражением сердца, почек, тяжелой анемией и неврологическими расстройствами, умершей через две недели от начала болезни. При аутопсийном исследовании был выявлен распространенный тромбоз сосудов микроциркуляторного русла (МЦР) – терминальных артериол и капилляров – разных органов. Типичная клиническая картина этого заболевания, отличающегося крайне высокой летальностью (более 90 % до начала использования для лечения свежезамороженной плазмы), характеризуется пентадой признаков: тяжелая тромбоцитопения, гемолитическая анемия, неврологические нарушения, ренальная дисфункция и лихорадка. Долгое время патогенез ТТП оставался неясным, и лишь в 1980-х гг. была установлена связь генерализованного тромбообразования в сосудах микроциркуляторного русла с присутствием в кровотоке сверхкрупных мультимеров фактора фон Виллебранда (vWF), обладающих избыточной активностью в отношении адгезии и агрегации тромбоцитов [1, 2]. Впоследствии двумя группами исследователей независимо
друг от друга в плазме был идентифицирован специфический энзим, расщепляющий сверхкрупные мультимеры vWF до более мелких, хотя и остающихся активными гемостазиологически, но не способных вызывать распространенное микротромбообразование [3, 4]. Им оказался ADAMTS13 (a disintegrin and metalloprotease with thrombospondin-1-like domains, member 13) – металлопротеаза, принадлежащая к семейству пептидазных белков ADAM, биологическая роль которых заключается в деградации экстрацеллюлярного домена трансмембранных белков. Дефицит или отсутствие активности этого фермента играет ключевую роль в патогенезе ТТП, приводя к накоплению в циркуляции сверхкрупных мультимеров vWF вследствие дефекта их деградации [3, 4]. Причинами дефицита ADAMTS13 могут быть многочисленные мутации гена, кодирующего синтез этой металлопротеазы (сегодня их известно более 70), или выработка антител к ней [4–6], в зависимости от чего выделяют два основных типа ТТП: наследственную (сидром Upshaw–Shulman) и приобретенную. Развитие последней возможно при системной красной волчанке (СКВ), в т. ч. при волчаночном нефрите (ВН), в результате дефицита ADAMTS13, обусловленного анти-ADAMTS13-антителами [7, 8].
Недавними исследованиями установлено, что без немедленной деградации ADAMTS13 сверхкрупные мультимеры vWF могут быстро накапливаться в участках повреждения эндотелия, привлекая туда большие количества тромбоцитов, формирующих тромбы [9, 10], поскольку адгезивная активность vWF коррелирует с размером его мультимеров. Таким образом, ADAMTS13 регулирует функциональную
активность vWF, способствуя ограничению роста тромбов в микроциркуляторном русле.
Таблица 1. Распределение генотипов полиморфных маркеров генов гемостаза у обследованных больных.
Многочисленные исследования показали, что активность ADAMTS13 < 5 % вплоть до ее полного отсутствия (тяжелый дефицит металлопротеазы) характерна для ТТП [4, 6, 11, 12, 13]. Однако небольшое или умеренное снижение активности ADAMTS13, как оказалось, может наблюдаться при ряде заболеваний, в т. ч. кардио- и цереброваскулярных [14–16], циррозе печени, алкогольном гепатите, остром панкреатите [17, 18], сепсисе и индуцированном им синдроме диссеминированного
внутрисосудистого свертывания (ДВС) [19–21], а также при других микроангиопатических синдромах: гемолитико-уремическом синдроме (ГУС), злокачественной артериальной гипертонии [22–24] и антифосфолипидном синдроме (АФС) [25, 26]. Низкая активность ADAMTS13 была обнаружена
даже при гломерулярных поражениях почек разного генеза – волчаночном нефрите (ВН) и хроническом гломерулонефрите (ХГН) [12, 27]. Результаты этих исследований дают основания рассматривать ADAMTS13 как фактор формирования микроциркуляторных тромбозов не только при ТТП, но и при других микроангиопатических синдромах, а также гломерулярной патологии иммуновоспалительного генеза.
В последние годы получены данные, свидетельствующие о возможности развития ренальной тромботической микроангиопатии (ТМА) при наиболее частых генетических формах тромбофилии – Лейденской мутации V фактора свертывания крови, мутациях генов протромбина, ингибитора активатора плазминогена (PAI-1) и метилентетрагидрофолат-редуктазы (MTHFR) [28, 29]. Так, Т. Raife и соавт. обнаружили, что среди пациентов с клиническими и морфологическими признаками ТМА частота выявления мутации фактора V Leiden была достоверно выше по сравнению с контрольной группой, а в
исследованиях Sucker и соавт. выявлена связь между развитием ГУС/ТТП и носительством генотипа Т/Т гена MTHFR C 677 T и генотипа G/G гена тромбин-активируемого ингибитора фибринолиза (TAFI G505A) [30].
В наших недавних исследованиях установлено, что ТМА у пациентов с генетическими тромбофилиями аналогично нефропатии, ассоциированной с АФС (АФСН), может либо развиваться как единственная форма поражения почек, либо сочетаться с уже существующей нефропатией, чаще всего ХГН [31].
Однако работы по изучению роли ADAMTS13 и vWF в развитии тромбозов микроциркуляторного русла почек, ассоциированных с приобретенными и наследственными тромбофилиями, отсутствуют.
Целью нашего исследования было оценить состояние активности системы “ADAMTS13–фактор фон Виллебранда–тромбоциты” у пациентов с приобретенной (АФС) и наследственными тромбофилиями и поражением почек, либо развившимся как локально почечное проявление тромбофилии, либо сочетающимся с ХГН или ВН.
Материал и методы
В исследование были включены 20 больных с тромбофилиями и признаками поражения почек (средний возраст – 30 ± 11 лет): 14 (70 %) мужчин и 6 (30 %) женщин, находившихся в клинике им. Е.М. Тареева в 2007–2010 гг., и 14 здоровых добровольцев, сопоставимых по полу и возрасту. Критерием отбора пациентов было наличие морфологических признаков ТМА в нефробиоптатах. На момент выполнения биопсии почки оценивали основные клинико-лабораторные проявления нефропатии: артериальное давление (АД), суточную протеинурию (СПУ), эритроцитурию, скорость клубочковой фильтрации (СКФ) методом Реберга–Тареева, креатинин сыворотки (Scr), уровень альбумина крови.
Критериями артериальной гипертонии (АГ) были уровень систолического АД (САД) ≥ 140 мм рт. ст. и/или диастолического АД (ДАД) ≥ 90 мм рт. ст. до назначения антигипертензивных препаратов. При уровне АД ≥ 180/110 мм рт. ст. АГ расценивали как тяжелую. Нарушение функции почек констатировали при СКФ ниже 80 мл/мин и/или уровне Scr выше 1,4 мг/дл. “Почечным исходом” считали стабильное повышение уровня Scr > 1,4 мг/дл в течение не менее 6 месяцев. Протеинурию определяли как суточную экскрецию белка не менее 0,1 г/сут (при значениях 0,1–0,4 г/сут – минимальная, при 0,5–3,0 г/сут – умеренная, более 3 г/сут – выраженная). Гематурию определяли как умеренную при количестве эритроцитов от 4 до 20 в поле зрения, как выраженную – более 20 в поле зрения.
Длительность почечного анамнеза определяли на основании медицинской документации, в которой имелись данные об отсутствии поражения почек за относительно небольшой период времени до появления признаков нефропатии, и рассчитывали в месяцах от дебюта болезни до момента биопсии
почек. Средняя длительность почечного анамнеза к моменту проведения биопсии составила 10 [2; 96] месяцев.
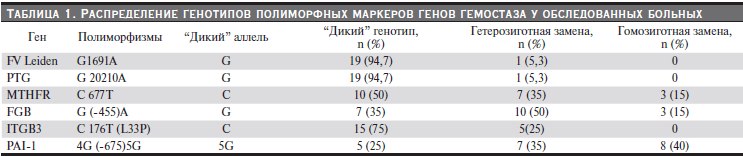
У всех больных определяли серологические маркеры АФС (антитела к кардиолипину, волчаночный антикоагулянт, антитела к β2-гликопротеину I) и полиморфизмы следующих генов свертывающей системы крови: метилентетрагидрофолатредуктазы С677Т (MTHFR (C677T), протромбина G20210A
(PTG (G20210A), фактора V Leiden G1691A (F5 (G1691A), β-цепи фибриногена -455G > A (FGB (-455G > A), тромбоцитарного гликопротеин IIIa T176C, L33P (ITGB3 (T176C, L33P) и ингибитора активатора плазминогена I типа -675 4G/5G (PAI-1 (-675 4G/5G) методом полимеразной цепной реакции (ПЦР).
У 5 (25 %) больных диагностирован АФС: первичный – у 1, вторичный при СКВ – у 4 пациентов. У 15 (75 %) больных выявлена мультигенная тромбофилия, представленная различными сочетаниями “протромбогенных” генотипов исследованных генов (табл. 1).
Для определения уровня и активности vWF, а также активности ADAMTS13 в плазме крови венозную кровь смешивали с антикоагулянтом (цитрат натрия), после чего центрифугировали при ускорении 3000 g в течение 20 минут и замораживали при температуре -20 °С. Активность ADAMTS13 определяли методом FRET (fluorescence resonance energy transfer) с использованием флюорогенного субстрата FRETS-VWF73 (PeptaNova GmbH, Germany) и выражали в процентах (%). Интервал активности ADAMTS13 у здоровых составил 94–113 %.
Концентрацию vWF в плазме крови (vWF:Ag) и его активность определяли методом твердофазного иммуноферментного анализа (ELISA) с помощью поликлональных антител (Technozym vWF:Ag ELISA; American diagnostic inc. vWF activity kit). Уровень vWF у здоровых волонтеров составил 0,5–1,5 U/ml, а активность vWF – 56 % [31;83,5].
Для морфологического исследования использовали материал, полученный с помощью чрескожной пункционной биопсии почки. В биоптатах оценивали частоту и характер гломерулярных изменений, изменений интерстиция, канальцев и сосудов, наличие и особенности иммунных отложений, гломерулярного склероза.
При статистической обработке полученных данных рассчитывали среднее значение и стандартное отклонение (mean ± SD) или медиану, 25-й и 75-й квартили – Me [25 %, 75 %], в зависимости от соответствия данных нормальному распределению). Для проверки статистической значимости различий частотных показателей использовали критерий χ2 по Пирсону. Достоверными считались различия при р < 0,05; 0,05 < р < 0,1 рассматривали как тенденцию к различию.
Для выявления и оценки связей между исследуемыми показателями применялся непараметрический корреляционный анализ по методу Spearman. О силе и направленности связи судили по величине и знаку коэффициента регресса r. Достоверными считали различия при р < 0,05. Статистическую
обработку данных выполняли с помощью пакета программ SPSS 11,5.
Результаты
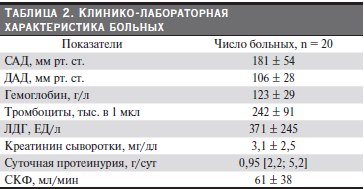
Клиническая картина поражения почек представлена нефротическим синдромом у 7 (35 %) больных, остронефритическим синдромом – у 3 (15 %), изолированным мочевым синдромом – у 10 (50 %) пациентов. Протеинурия (ПУ) выявлена у 18 больных, у 10 (55,6 %) из которых носила изолированный характер. Величина ПУ составила 0,95 [2,2; 5,2] г/сут. Эритроцитурия зарегистрирована у 16 (80 %) пациентов, в т. ч. выраженная – у 6. Нарушение функции почек отмечено у 15 больных. Все они
имели сниженную СКФ, средняя величина которой составила 61 ± 38 мл/мин, у 12 (75 %) из них снижение СКФ сочеталось с повышением уровня Scr. У 2 (10 %) больных развилась ОПН, потребовавшая лечения сеансами гемодиализа. “Почечный исход” наступил у 10 больных.
Клинико-лабораторная характеристика больных представлена в табл. 2.
Анемия выявлена у 8(40 %) больных, значения гемоглобина у которых составили 55–116 г/л. Повышение уровня ЛДГ от 480 до 834 МЕ имели 4 (20 %) из этих пациентов. Тромбоцитопения
(число тромбоцитов – 53 тыс. в 1 мкл крови) отмечена лишь у 1 (5 %) пациентки, имевшей наиболее низкий показатель гемоглобина и высокий – ЛДГ. Таким образом, лабораторные признаки ТМА в виде тяжелой гемолитической анемии и выраженной тромбоцитопении зарегистрированы только в одном
случае – у больной СКВ. Еще у трех пациентов констатирована изолированная микроангиопатическая гемолитическая анемия с умеренными снижением уровня гемоглобина и повышением ЛДГ при нормальном числе тромбоцитов. Тромбозы развились у 8 (40 %) больных: артериальные – у 5, венозные – у 2, смешанные – у 1 больного.
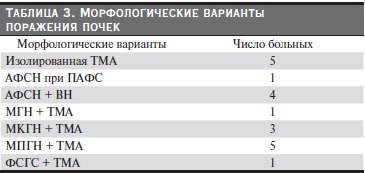
У всех больных в биоптатах почек имелись морфологические признаки ТМА, представленные гломерулярным и/или артериолярным тромбозом, утолщением интимы артерий с клеточной пролифераций по типу “луковичной шелухи”, утолщением базальных мембран капилляров клубочков, артериолосклерозом, интерстициальным фиброзом, атрофией канальцев, ишемической атрофией коры. При этом у 5 (25 %) больных мультигенной тромбофилией ТМА оказалась единственным вариантом поражения почек; у 1 больного была проявлением АФС-нефропатии при первичном АФС; у 4 с вторичным АФС сочеталась с волчаночным нефритом (ВН); у 10 пациентов ТМА сочеталась с различными морфологическими формами ХГН: у 3 – с мезангиокапилярным ГН, у 5 – с мезангиопроли-
феративным ГН, у 1 – с мембранозным ГН, у 1 – с фокально-сегментарным гломерулосклерозом (табл. 3).
Обращали на себя внимание частота и выраженность склеротических изменений в ткани почек при относительно коротком анамнезе нефропатии. Сегментарный или глобальный гломерулосклероз выявлен у 12 (60 %) больных, артерио- и/или артериолосклероз – у 17 (85 %), склероз интерстиция у 15 (75 %) больных. В 15 биоптатах имелись изменения базальных мембран клубочков (БМК), представленные утолщением различной степени выраженности и/или удвоением независимо от морфологического варианта нефропатии. Полнокровие капилляров отмечено у 10 (50 %)больных. Признаки острой ТМА обнаружены у 2 (10 %) больных: у одной с СКВ, имевшей признаки микроангиопатического гемолиза и тромбоцитопению в сочетании с ВН III класса, у другого – с мембранозным ГН. При иммуногистохимическом исследовании отложения фибриногена по ходу БМК, в стенках артериол и капилляров были выявлены у 10 больных.
Рисунок 1. Связь активности ADAMTS13 с уровнем vWF.
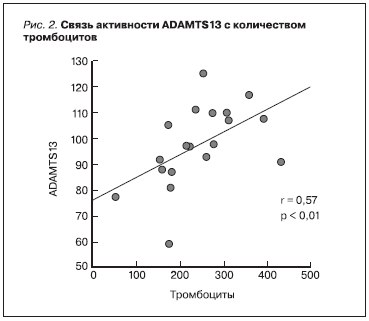
Рисунок 2. Связь активности ADAMTS13 с количеством тромбоцитов.
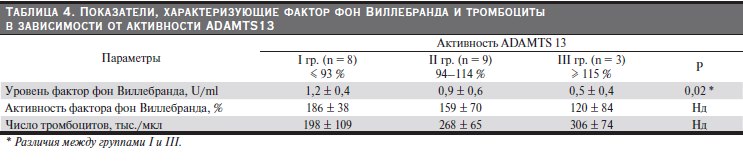
Активность ADAMTS13 в группе контроля составила 94–114 %, в группе больных – 56–123 % (р > 0,1). В зависимости от активности ADAMTS13 у здоровых больные были разделены на 3 группы: с активностью ниже крайнего референсного значения, в пределах референсных значений и выше них. Активность ADAMTS13 ниже крайнего референсного значения (< 94 %), составившая 92–56 %, обнаружена у 8 (40 %) больных (табл. 4), из которых 2 страдали СКВ, в одном случае с АФС, 4 – ХГН, 1 – первичным АФС и 1 имел только генетическую тромбофилию. У тех же пациентов отмечены наиболее высокие показатели уровня и активности vWF, а также наименьшее количество тромбоцитов. Последнее хотя и было в пределах нормальных значений, тем не менее оказалось значительно ниже, чем у больных II и III групп. В III группе, в которой активность ADAMTS13 превышала референсные значения, исследованные показатели имели обратные значения: уровень и активность vWF оказались наиболее низкими, а число тромбоцитов – наиболее высоким. Во II группе эти показатели имели промежуточные значения (табл. 4). Корреляционный анализ выявил обратную зависимость между активностью ADAMTS13 и уровнем ФВ
(r = -0,3, р < 0,05) (рис. 1), прямую – между активностью ADAMTS13 и числом тромбоцитов (r = 0,57, р < 0,01) (рис. 2). Наиболее низкие показатели активности ADAMTS13 (56 и 76 %) отмечены у двух больных с гистологической картиной острой ТМА в биоптатах почек, у одной из которых она сочеталась с III классом ВН, у другого – с мембранозной нефропатией.
Таблица 4. Показатели, характеризующие фактор фон Виллебранда и тромбоциты в зависимости от активности ADAMTS13.
Поскольку значения ADAMTS13, превышающие референсные, не ассоциированы с развитием патологии, мы объединили пациентов, у которых значения металлопротеазы находились в пределах референсных или были выше них, в единую группу с “нормальными” показателями активности ADAMTS13. При сравнении основных характеристик почечного процесса в этой группе (12 больных) и в группе больных со сниженными показателями ADAMTS13 (8 больных) оказалось, что в отсутствие
различий в величине протеинурии (3,5 ± 6,0 vs 3,1 ± 3,8 г/сут, р > 0,05), САД (188,7 ± 57,4 vs 159,0 ± 39,8 мм рт. ст., р > 0,05) и ДАД (109,3 ± 30,0 vs 98,0 ± 21,7 мм рт. ст., р > 0,05) у пациентов с “нормальным” ADAMTS13 СКФ была выше (47,9 ± 28,9 vs 37,9 ± 30,2 мл/мин), а уровень креатинина сыворотки ниже, чем у больных с “низким” ADAMTS13 (2,5 ± 1,5 vs 3,7 ± 2,7 мг/дл, р = 0,07). “Почечного исхода” достигли 5 из 8 (62,5 %) пациентов со сниженным уровнем металлопротеазы и 5 из 12
(41,6 %) – с “нормальным” (р > 0,05).
Обсуждение
В проведенное исследование были включены пациенты с приобретенной (АФС) и наследственной тромбофилиями и поражением почек, гистологические признаки которого во всех случаях соответствовали диагнозу ТМА, изолированной у 6 больных (у 1 – с первичным АФС и 5 – с мультигенной формой тромбофилии) и сочетающейся с ВН у 4 пациентов с вторичным АФС или различными морфологическими вариантами ХГН – у 10, оказавшихся носителями нескольких “протромбогенных” полиморфизмов генов гемостаза. Таким образом, результаты настоящей работы подтверждают установленную нами ранее возможность развития ТМА у больных с мультигенной тромбофилией либо как единственной формы поражения почек аналогично возможности возникновения изолированной АФС-ассоциированной нефропатии при АФС [32], либо в сочетании с другими гломерулярными болезнями, главным образом с ХГН [33].
Несмотря на несомненные морфологические признаки ТМА, лишь у одной пациентки с волчаночным нефритом и быстронарастающим уровнем креатинина сыворотки выявлены тромбоцитопения и микроангиопатическая гемолитическая анемия, позволяющие предполагать развитие ТТП, хотя
неврологические расстройства и лихорадка отсутствовали; еще у трех больных признаки микроангиопатического гемолиза отмечены в отсутствие тромбоцитопении. Таким образом, среди всех больных, включенных в исследование, клиническая картина была представлена преимущественно признаками поражения почек с развитием ренальной дисфункции у 75 % из них, тогда как гематологические проявления ТМА отсутствовали у большинства пациентов. Ни в одном случае не был
установлен диагноз ТТП.
Наши данные совпадают с недавно опубликованными результатами исследования S.A. Serres и P. Isenring, обнаруживших ТМА в биоптатах почек больных с неясными причинами нарушения функции почек (что и послужило показанием к выполнению нефробиопсии), у которых отсутствовала тромбо-
цитопения, а у части из них – и анемия. Авторы назвали такую форму ТМА “атромбоцитопенической” и предположили ее возможную связь с нарушениями ADAMTS13 и vWF, которые, однако, в данной работе не исследовались [34].
Это предположение подтвердили результаты нашего исследования, обнаружившего снижение активности ADAMTS13 наряду с повышением уровня и активности vWF у 40 % больных с приобретенной (АФС) и наследственной тромбофилиями при наличии ТМА независимо от того, существовала ли она изолированно, или сочеталась с ХГН или ВН. Несмотря на нормальное число тромбоцитов в группе больных со сниженной активностью ADAMTS13, мы, как и другие авторы, обнаружили прямую связь между этими двумя показателями и обратную – между активностью ADAMTS13 и уровнем vWF [24], что позволяет обсуждать возможность потребления тромбоцитов в процессах тромбообразования в МЦР почек и при умеренном дефиците ADAMTS13. Даже небольшое снижение активности этой металлопротеазы, по-видимому, может приводить к неполной деградации и, следовательно, накоплению сверхкрупных мультимеров vWF, играющих ключевую роль в образовании тромбоцитарных агрегатов, которые составляют основу тромбов в сосудах микроциркуляторного русла [35]. Наши данные согласуются с результатами других исследований, в которых незначительный или
умеренный дефицит ADAMTS13 был обнаружен у больных ГУС/ТТП, ассоциированных с беременностью и родами, приемом различных лекарств, трансплантацией костного мозга, сепсисом, опухолями, злокачественной артериальной гипертензией, а также у больных постдиарейным ГУС [23, 24, 36, 37]. Неожиданными оказались данные S.K. Vessly и соавт., установивших, что почти у двух третей пациентов с идиопатической ТТП активность ADAMTS13 была более 5 % , причем у 36 % больных превышала 25 % [36]. Эти данные легли в основу предположения, будто умеренный дефицит
ADAMTS13 (> 10 %) можно рассматривать как дополнительный фактор риска микроциркуляторных тромбозов различного генеза [37] даже при ТТП. Его правомерность подтверждают наблюдения позднего дебюта наследственной ТТП, впервые развившейся у взрослых [38]. Причина отсроченного начала болезни неясна, но острому эпизоду, как правило, предшествуют инфекции, беременность,
хирургические операции, т. е. состояния, при которых развивается дисфункция эндотелия, сопровождающаяся секрецией сверхкрупных мультимеров vWF, что дает основания предполагать необходимость триггерного механизма ТМА даже при наследственном дефиците ADAMTS13, особенно
если он выражен умеренно [24, 36, 37].
Умеренный дефицит ADAMTS13 может быть обусловлен повышенным потреблением, уменьшенным синтезом или прямой ингибицией [37]. Установлено, что некоторые провоспалительные цитокины (фактор некроза опухоли-α, интерлейкин-4, интерферон-γ) могут ингибировать транскрипцию
этой металлопротеазы в звездчатых клетках печени и эндотелиоцитах и ее секрецию из них [39], а также непосредственно блокируют активность ADAMTS13 в кровотоке [40]. В связи с этим оправданно предположение об опосредованных цитокинами локально почечных подавлениях секреции ADAMTS13
или прямой его блокаде у больных активными формами волчаночного или брайтова нефрита в нашем исследовании. Возможно, именно поэтому в системной циркуляции отмечено лишь незначительное снижение активности металлопротеазы. С другой стороны, эндотелиальная дисфункция, характерная
для ХГН и особенно ВН, может проявляться повышением уровня и активности vWF в плазме крови, как было установлено ранее И.Н. Бобковой и соавт. [41]. Повреждение клеток эндотелия может быть вызвано также воздействием антифосфолипидных антител, что объясняет рост активности vWF при АФС-нефропатии. Таким образом, у пациентов с ВН, ХГН или АФСН, имеющих морфологические признаки ТМА, возможен дисбаланс между активностью ADAMTS13 и vWF, который, как полагают, более важен, чем абсолютные концентрации этих факторов в кровотоке [24]. При этом взаимодействие между ними может осуществляться двояко: с одной стороны, освобождение избыточных количеств vWF из поврежденных эндотелиоцитов может связывать ADAMTS13, приводя к уменьшению его активности, с другой – сниженная активность ADAMTS13 способствует экспрессии сверхкрупных мультимеров vWF, обладающих наиболее высокой протромбогенной активностью [24], что в обоих случаях будет индуцировать тромбообразование в сосудах МЦР почек с развитием ишемии органа. По-видимому, имевшуюся у большинства наших больных с умеренным дефицитом ADAMTS13
мультигенную тромбофилию, представленную сочетанием нескольких “протромбогенных” генотипов генов гемостаза, можно считать дополнительным фактором, вносящим вклад в развитие микроциркуляторных тромбозов за счет избыточной активации внутриклубочкового свертывания крови, не адекватной выраженности иммунного воспаления, как было установлено в нашем предыдущем исследовании [31]. Полученные нами данные свидетельствуют о справедливости теории “двойного удара” применительно к развитию микроциркуляторных тромбозов, которые у больных ВН, ХГН и АФСН, по-видимому, обусловлены несколькими факторами, в т. ч. активностью иммуновоспалительного процесса, генетическими нарушениями в системе гемостаза и в ряде случаев –
дефицитом ADAMTS13. Сочетание перечисленных факторов скорее всего способно обеспечивать прогрессирующее течение почечного процесса, в пользу чего свидетельствуют более выраженное повышение креатинина сыворотки у пациентов с дефицитом ADAMTS13 по сравнению с больными, имевшими нормальную его активность, а также большая частота достижения “почечного исхода” при сниженной активности металлопротеазы. Наши данные согласуются с результатами исследований, в которых установлена обратная связь между сывороточным креатинином и активностью ADAMTS13 у
больных злокачественной артериальной гипертензией и ДВС-синдромом при сепсисе, что дало основания авторам этих работ предполагать важную роль даже небольшого дефицита ADAMTS13 в развитии органного повреждения при различных микроангиопатических синдромах, сопровождающихся активацией и дисфункцией эндотелия [20, 24].
Заключение
Таким образом, результаты нашего исследования позволяют рассматривать нарушения в системе “ADAMTS 13–vWF–тромбоциты” как один из универсальных патогенетических механизмов тромбообразования в сосудах микроциркуляторного русла почек при различных видах патологии и обосновывают возможность развития ТМА при иммуновоспалительных заболеваниях почек (ВН, ХГН) и АФС-нефропатии. Носительство множественных полиморфизмов генов гемостаза, как и антифосфолипидные антитела, по-видимому, служит фактором, предрасполагающим не только к артериальным и/или венозным, но и микроциркуляторным тромбозам, и в ряде случаев может играть роль локального триггерного механизма гиперкоагуляции.
Исследование проводится при поддержке РФФИ (грант № 09-04-00468-а).



{kind=link}
{kind=link}
{kind=link}
{kind=link}