
Хроническая болезнь почек (ХБП) — доказанный фактор риска развития и прогрессирования сердечно-сосудистых осложнений, сопровождающийся рядом метаболических нарушений [1]. В числе последних — нарушение липидного обмена.
Изменения метаболизма липидов возникают уже на ранних стадиях почечного поражения при снижении скорости клубочковой фильтрации (СКФ) до 50 мл/мин [2] и усиливаются по мере прогрессирования ХБП. По нашим данным, среди 165 больных со 2-4-й стадиями ХБП гиперлипидемия выявлялась в 80 % случаев [3]. Спектр дислипидемии при почечной недостаточности отличается от такового по сравнению с общей популяцией. Выявляются изменения во всех классах липопротеинов, варьирующие в зависимости от стадии ХБП, применяемых методов лечения и приема лекарственных препаратов, влияния модифицируемых конкурирующих заболеваний и состояний, таких как сахарный диабет (СД) и/или нефротический синдром (НС) [4, 5].
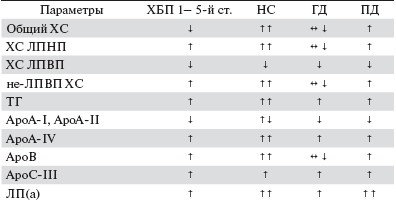
Профиль липидов и липопротеинов плазмы при ХБП
Одним из частых проявлений нарушений липидного обмена при ХБП является повышение уровня триглицеридов (ТГ). Гипертриглицеридемия сопряжена с ростом концентрации и снижением клиренса липопротеинов очень низкой плотности (ЛПОНП) и аккумуляцией липопротеинов промежуточной плотности (ЛППП). При ХБП снижается также клиренс хиломикрон (ХМ) с накоплением их ремнант. Значения липопротеинов низкой плотности (ЛПНП) обычно в пределах нормы и очень редко повышаются в стадию терминальной хронической почечной недостаточности (ТПН). Уровень липопротеинов высокой плотности (ЛПВП), как правило, снижен. Концентрация холестерина (ХС) сыворотки обычно в пределах нормальных значений или даже снижается у больных ТПН [4-6].
При снижении почечной функции наряду с количественными нарушениями отмечаются качественные изменения в составе липопротеинов. Так, в ЛПОНП повышается относительное содержание ХС и снижается - ТГ, тогда как для ЛПНП характерны прямо противоположные изменения. В ЛПВП снижена концентрация свободного и этерифицированного ХС, но повышена концентрация ТГ [4-6]. Вышеописанные липидные сдвиги отмечаются почти у всех больных с умеренным и выраженным нарушением функции почек (даже при нормальном уровне в плазме общего ХС и ТГ). Нередко развитие и прогрессирование ХБП сопровождаются выраженной ПУ, при повышении общего ХС и ХС ЛПНП [7]. Однако по мере усугубления ХБП вплоть до стадий, сопряженных со снижением скорости клубочковой фильтрации и, соответственно, снижения ПУ, липидный профиль становится типичным для ХБП.
Наряду с изменением количества и перераспределением липидов уремия оказывает влияние и на содержание аполипопротеинов: характерно снижение плазменных концентраций ароА-1, ароА-2 и апоЕ, повышение ароС-III и изменение соотношения апоСII/апоС-III [4, 5, 8]. Спектр возможных липидных изменений при ХБП представлен в табл. 1.
Таблица 1. Липидный профиль при поражении почек.
Примечание. Не-ЛПВП ХС включает ХС ЛПНП, ЛПОНП, ЛППП, ХМ и их ремнант; норма(↔), повышение (↑), снижение (↓).
Механизмы развития уремической дислипидемии
Изменения в метаболизме ЛПВП
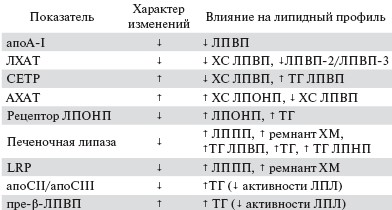
Индуцированные ХБП липидные нарушения приводят к снижению ХС ЛПВП, нарушению превращения субфракций ЛПВП-3 в ЛПВП-2, повышению ТГ ЛПВП и снижению в плазме уровня ароА-1. Нарушается обратный транспорт ХС от периферических тканей к печени. Все это приводит к нагрузке сосудистой системы холестерином и способствует атерогенезу [4, 5]. Обсуждаются различные механизмы нарушения метаболизма ЛПВП.
Лецитин-холестерин-ацетилтрансфераза (ЛХАТ)
ЛХАТ служит основной детерминантой образования и содержания ЛПВП в плазме крови. У пациентов с ТПН ее активность снижена [9]. Дефицит данного фермента может приводить к снижению уровня ЛПВП в плазме и нарушениям превращения ЛПВП при ХПН, что сопровождается значительным повышением свободного сывороточного ХС и снижением концентрации эстерифицированного ХС. В эксперименте на крысах с уремией уменьшение экспрессии гена ЛХАТ сопровождалось параллельным снижением содержания и активности этого фермента в плазме [10].
Белок-транспортер эфиров ХС (СЕТР)
Осуществляет перенос эстерифицированного ХС из ЛПВП к ЛППП в обмен на ТГ. Считают, что увеличение концентрации СЕТР может приводить к снижению эстерифицированного ХС в ЛПВП и увеличению ТГ ЛПВП. Почти у трети больных, находящихся на гемодиализе (ГД), выявляется повышение уровня СЕТР в плазме крови [11], однако механизм этого явления не ясен. У больных с выраженной протеинурией в сочетании с начальной и умеренной степенью нарушения почечной функции отмечалось усиление синтеза и повышение концентрации белка-переносчика в плазме крови [12].
Печеночная липаза
Катализирует гидролиз и возврат ТГ, содержащихся в ЛПВП. В экспериментальных и клинических работах выявлен дефицит этого фермента при ХБП, что может приводить к повышению содержания ТГ в ЛПВП [13].
Аполипопротеины ароА-I и ароА-II
Аполипопротеины ароА-Iи ароА-II входят в состав ЛПВП. АроА-I является активатором ЛХАТ и лигандом к рецепторам ЛПВП, апоА-II — активатором печеной липазы. У больных ТПН концентрации этих апопротеинов значительно снижены [14]. В эксперименте индуцированное ХПН уменьшение содержания ароА-Iв плазме было следствием снижения экспрессии гена ароА-I в печени [15].
Тканевой рецептор к ЛПВП (SRB-1)
Печеночный рецептор для эфиров ХС и ТГ из ЛПВП. На экспериментальных животных при выраженной ПУ продемонстрировано снижение его экспрессии в печени [16], тогда как ХБП per se не влияла на активность печеночной мРНК [15]. Тем не менее авторы считают, что сочетание выраженной ПУ и ХБП может влиять на экспрессию этого рецептора и следовательно, на обратный транспорт ХС.
Ацетил-холестерин ацетилтрансфераза (АХАТ)
АХАТ — основной энзим внутриклеточной эстерификации ХС. Повышение ее активности снижает содержание ХС ЛПВП в плазме и нарушает созревание ЛПВП. Наличие ХБП вызывало активацию мРНК АХАТ-2 в печени с усилением синтеза и общей активности фермента [17]. При экспериментальной уремии использование ингибиторов АХАТ приводило к улучшению показателей липидного профиля и повышению клиренса креатинина, что, по мнению исследователей, связано с усилением процессов обратного транспорта ХС и может затормозить прогрессирование гломерулосклероза [18].
Важным компонентом ЛПВП является параоксоназа — фермент, ингибирующий окисление ЛПНП. При ХБП ее активность в плазме снижена, что способствует окислению ЛПНП и, возможно, ЛПВП [19]. Воспаление, ассоциированное с инфекцией или уремией, может способствовать превращению ЛПВП из антиоксидантных в прооксидантные частицы, что влияет на развитие атеросклероза при ХБП [20, 21]. Другим последствием хронического системного воспаления является гипоальбуминемия. Дефицит альбумина, являющегося переносчиком свободного ХС от периферических тканей к ЛПВП, может приводить к снижению уровня ЛПВП [22].
Изменения в метаболизме ТГ
Уровень ТГ плазмы крови повышается уже на ранних стадиях ХБП, достигая максимальных значений у больных с НС и у пациентов, получающих заместительную почечную терапию [5]. Происходит накопление ТГ, представленных ХМ и ЛПОНП, с аккумуляции высокоатерогенных ремнантных частиц (ремнантные ХМ и ЛППП). В ЛОНП повышается содержание ХС и снижается ТГ, тогда как в ЛПНП отмечаются противоположные соотношения (повышение ТГ ЛПНП, снижение ХС ЛПНП) [4, 5, 23]. Среди возможных механизмов уремической гипертриглицеридемии обсуждают различные процессы: дизрегуляцию экспрессии/активности ферментов, нарушения в рецепторном аппарате, снижение метаболизма ЛПВП. Почечная недостаточность может вызывать инсулинорезистентность (ИР) [24], которая в свою очередь усиливает продукцию ЛПОНП в печени, что частично объясняет гипертриглицеридемию [5, 25]. Из-за снижения активности липаз также нарушается расщепление ТГ до свободных жирных кислот, необходимых для обеспечения энергетических потребностей организма, что может оказывать влияние на развитие синдрома белково-энергетической недостаточности (БЭН) у больных, находящихся на ГД [26].
Липопротеинлипаза (ЛПЛ)
При ХБП происходит подавление генной экспрессии ЛПЛ в жировой ткани, скелетных мышцах и миокарде, при этом происходит снижение ее активности [27]. Назначение гепарина в ходе сеансов ГД приводит к истощению эндотелиального пула ЛПЛ, что было продемонстрировано у этого контингента больных [28]. Определенную роль отводят ингибиторам активности фермента, ассоциированным с уремией: повышенной концентрации пре-β-ЛПВП, наличию ИР, вторичному гиперпаратиреозу [4, 26, 29, 30]. Еще один механизм снижения активности ЛПЛ при ХБП — это дефицит аполипопротеинов. В ЛПОНП и ХМ снижается содержание апоС-II (кофактора ЛПЛ) и апоЕ (лиганда для связывания с эндотелием) [31], что в дальнейшем приводит к нарушению превращения субфракций ЛПВП-3 в ЛПВП-2.
Печеночная липаза (ПЛ)
ПЛ принадлежит центральная роль в метаболизме липопротеидов, т. к. она участвует в гидролизе ТГ ЛППП с образованием ЛПНП, гидролизе ТГ и фосфолипидов в ЛПВП и ремнантах ХМ. ХБП вызывает снижение генной экспрессии и активности ПЛ [13], что влияет на метаболизм ремнант липопротеинов и ЛПВП. Дефицит этого фермента может возникнуть из-за вторичного гиперпаратиреоза или нарушения регуляции Са2+ в цитозоле [13].
Белок, подобный рецептору ЛПНП
Этот белок служит рецептором для ремнант ХМ и ЛППП в печени. В эксперименте на крысах с ХБП была снижена экспрессия гена данного рецептора, что привело к накоплению в плазме атерогенных ремнант [32].
Рецепторы к ЛПОНП
При экспериментальной почечной недостаточности было выявлено снижение экспрессии мРНК и нарушение образования рецепторов к ЛПОНП в жировой ткани, скелетных мышцах и миокарде у животных [33, 34]. Следствием этого является повышение концентрации ЛПОНП со снижением их клиренса, что способствует гипертриглицеридемии.
При уремии повышается содержание наиболее атерогенных ЛППП и мелких плотных частиц ЛПНП. Последние обладают высокой способностью пенетрировать в сосудистую стенку, окисляться и быть триггерами атеросклеротического процесса [4]. При использовании изотопных методов исследования показано, что у больных, леченных ГД, время нахождения в плазме ЛПНП и ЛППП более чем в 2 раза больше по сравнению со здоровыми людьми [35, 36]. При этом в результате процессов окисления, карбамилирования и гликирования происходит модификация их основного апоВ, что приводит к нарушению узнавания и связывания этих липопротеидов с рецепторами в печени и следовательно, к снижению их клиренса физиологическим путем [35].
Снижение катаболизма маскируется уменьшением образования ЛПНП, из-за чего в плазме определяется практически нормальный уровень ЛПНП [35]. Кроме того, модифицированные частицы ЛПНП активно захватываются скаверендж-рецепторами макрофагов, что приводит к накоплению ХС и образованию пенистых клеток, способствуя атерогенезу [4, 26, 36].
Наличие почечного повреждения — фактор, оказывающий влияние на сывороточный уровень ЛП(а). У больных с крупными изоформами апо(А), отмечалось повышение ЛП(а) уже на 1-й стадии ХБП — еще до снижения СКФ [37]. Данная связь прослеживалась во многих, но не во всех исследованиях, включавших больных ХБП без НС, а также пациентов, леченных ГД [38—42]. В то же время у больных НС [43] или находящихся на перитонеальном диализе [38], отмечено повышение всех изоформ ЛП(а) в плазме, возможно, как следствие выраженной потери белка с последующим усилением его продукции в печени [44]. Трансплантация почек приводит к снижению уровня крупных изоформ ЛП(а) у больных, ранее получавших лечение ГД [45], и всех изоформ ЛП(а) у больных, находившихся на перитонеальном диализе [46]. K. Frischmann et al. (2007) изучали метаболизм ЛП(а) у больных, леченных ГД, по скорости образования/катаболизма аполипопротеинов (апоА и апоВ), входящих в его состав [47]. Было показано, что у больных, леченных ГД, по сравнению со здоровым контролем скорость катаболизма ЛП(а) была значительно снижена с удлинением времени циркуляции апоА в кровотоке, при одинаковой скорости образования аполипопротеинов в обеих группах. Синдромы недостаточного питания и системного воспаления были ассоциированы с высоким уровнем ЛП(а) у больных, получавших терапию ГД [41, 42, 48].
АпоА-IV участвует в обратном транспорте холестерина, влияет на активность ЛХАТ, липопротеинлипазы и транспорт эстерифицированного ХС от ЛПВП к ЛПНП. В проспективном 7-летнем исследовании повышение уровня ароА-IV независимо от значений базальной СКФ было предиктором прогрессирования первичных недиабетических поражений почек, определенного по удвоению креатинина или по необходимости проведения ЗПТ [49]. У больных, получивших диализную терапию, отмечено 2-кратное повышение его уровня в плазме по сравнению с общей популяцией [8, 38, 50]. Участие почек в метаболизме данного гликопротеина подтверждается обнаружением ароА-IV в почечных канальцах [51], а также снижением концентраций ароА-IV в плазме крови при наличии протеинурии [52].
В табл. 2 представлены изменения ключевых ферментов и рецепторов липидного метаболизма при почечной недостаточности.
Таблица 2. Механизмы нарушения липидного метаболизма при ХБП (Vaziri N., 2006).
В последние годы предпринимались попытки выявить взаимосвязь “почечных” факторов риска с липидными расстройствами. Показано, что при хроническом гломерулонефрите снижение функции почек оказывало влияние на все показатели липидного обмена, выявлялись гиперхолестерин- и гипертриглицеридемия, гипоальфахолестеринемия, повышение уровня липидов (ХС и ТГ) и апоВ в составе ЛПНП и ЛПОНП [53]. Длительность гломерулонефрита прямо коррелировала с содержанием апоВ ЛПНП, длительность АГ — с ХС ЛПОНП и ТГ ЛПОНП, а степень снижения фильтрационной функции была обратно пропорциональной уровню α-ХС плазмы. В группе пациентов, получавших β-адреноблокаторы, отмечено повышение значений ТГ.
В другом исследовании изучали влияние пола и возраста на показатели липидного обмена у 175 больных с додиализной стадией ХБП и 108 пациентов, находившихся на программном гемодиализе [54]. Авторы показали, что у женщин в консервативной и терминальной стадиях ХБП отмечено повышение уровня атерогенных фракций липопротеинов по сравнению с мужчинами с аналогичной степенью нарушения функции почек. Возраст оказывал влияние на обмен липопротеидов только у женщин.
Характер дислипидемии зависит от типа заместительной почечной терапии. У больных, леченных ГД, отмечается нормальный или близкий к этому уровень общего ХС и ХС ЛПНП. У 20—40 % пациентов выявляется повышение значений ТГ и снижение ХС ЛПВП, у трети больных — подъем концентрации окисленных ЛПНП и ЛП(а) [6, 23]. Гипертриглицеридемия возникает из-за наличия ЛПОНП, богатых ТГ. Использование высокопроницаемых полисульфонных или целлюлозных триацетатных диализных мембран сопровождалось значительным снижением уровня ТГ с повышением уровня ароА-I и ХС ЛПВП в сыворотке крови [55].
У больных, находившихся на перитонеальном диализе, отмечен более атерогенный липидный профиль по сравнению с пациентами, получавшими лечение сеансами ГД. У 20—40 % этих больных повышен общий ХС и ХС ЛПНП, у 25—50 % — уровень ТГ и апоВ, у 42 % больных — уровень окисленных ЛПНП и ЛП(а). Также повышены концентрации мелких плотных частиц ЛПНП. Значения ХС ЛПВП снижены. Гипертриглицеридемию при перитональном диализе связывают с усиленным синтезом ЛОНП в печени [6, 23]. Существующая ИР и поступление глюкозы из диализата усиливают липогенез de novo и образование ЛПОНП [6]. Увеличенный клиренс апопротеинов через брюшину также может быть триггером дислипидемии [6]. С потерей значительного количества белка с диализной жидкостью связывают более атерогенный характер липидных изменений при перитонеальном диализе.
Возможные механизмы влияния дислипидемии на почечное и сосудистое поражение у больных ХБП
Гиперлипидемия может реализовать свое влияние на прогрессирование почечного повреждения несколькими путями: способствуя развитию интраренального атеросклероза или через токсическое влияние липидов на структуры нефрона [8, 26]. Профильтровавшиеся СЖК, фосфолипиды и ХС реабсорбируются клетками канальцевого эпителия, что может стимулировать тубулоинтерстициальное воспаление, образование пенистых клеток и тканевое повреждение. ЛПНП активируют рецепторы, экспрессируемые мезангиальными клетками, стимулируют продукцию матриксных белков и провоспалительных цитокинов, хемокинов и факторов роста (МСР-1, ИЛ-6) и активацию макрофагов. Накопление липопротеинов в мезангии усиливает образование матрикса и способствует гломерулосклерозу [8, 26, 56]. В эксперименте гиперхолестеринемия и гипертриглицеридемия были ассоциированы с повреждением подоцитов, что также приводило к склерозу мезангия [57]. Дисбаланс между факторами, усиливающими/ ингибирующими процессы вазоконстрикции, коагуляции, клеточной пролиферации и трансдифференцировки, приводит к соответствующим почечным и сосудистым последствиям, что позволяет провести параллели между развитием гломерулосклероза и атеросклероза [58].
На стадии выраженной ХБП липопротеины подвергаются модификации и становятся окисленными ЛПНП (ок-ЛПНП). Ок-ЛПНП способствуют адгезии моноцитов к эндотелию капилляров клубочка и оказывают влияние на клетки канальцевого эпителия [59]. Они связываются с рецепторами в мезангии и через ряд клеточно-молекулярных механизмов усиливают в нем воспалительные и фиброгенные процессы. Цитотоксический эффект ок-ЛПНП проявляется в индукции апоптоза подоцитов с потерей нефрина и повреждением гломерулярного барьера. Обсуждается влияние других модификаций липопротеидов (гликированный и ацетилированный ЛПНП) в почечном и сосудистом повреждениях [8]. В эксперименте в патогенезе почечного поражения показана роль оксидативного стресса [60].
В эксперименте у мышей с дефицитом АроЕ (-/-), получавших высококалорийную диету, индуцировали почечную недостаточность. При этом отмечено нарастание уровней окисленных и карбамилированных ЛПНП. Уровень последних коррелировал с более выраженным сосудистым атеросклеротическим повреждением сосудов. Аккумулированные в стенке аорты карбамилированные ЛПНП в дальнейшем колонизировались макрофагами при участии IСАМ-1 и это позволяет рассматривать их как фактор риска развития атеросклероза при уремии [91].
Возможно, что некоторые из неблагоприятных эффектов липидов на почки опосредуются другими механизмами. Профиль липидных нарушений при ХПН схож с таковым у больных с синдромом ИР [25]. Можно предположить, что нарушение чувствительности тканей к инсулину может лежать в основе или опосредовать связь между липидными нарушениями и потерей почечной функции. При диабетической нефропатии ИР играет роль в сосудистом повреждении, т. к. она связана со многими нарушениями, включая высокое АД, липидные изменения, развитие гипертрофии миокарда [61].
Последствия нарушений липидного метаболизма при ХБП
Дислипидемия как фактор развития и прогрессирования ХБП
Впервые Moorhead et al. (1982) постулировали потенциальную связь между дислипидемией и поражением почек [62], что нашло подтверждение в дальнейших работах. Так, в ARIC Study развитие почечной дисфункции ассоциировалось с высоким уровнем ТГ и снижением ЛПВП [63]. В Physicians Health Study у здоровых лиц с исходно нормальной функцией почек изменения в липидном профиле, включившие повышение общего ХС, ХС не-ЛПВП, снижение ХС ЛПВП, коррелировали с риском повышения креатинина сыворотки крови [64]. Низкий уровень ХС ЛПВП был независимым фактором риска развития ХБП в Framingham Offspring study [65]. В проспективном 5-летнем наблюдении за 1428 больных с диабетом и СКФ > 70 мл/мин при наличии гиперхолестеринемии чаще выявлялось нарушение функции почек как у больных сахарным диабетом (СД), так и без него [66]. Аналогичная тенденция отмечена в ретроспективном исследовании пациентов с эссенциальной АГ и исходно нормальной СКФ, где базальные значения ХС и САД были идентифицированы как факторы риска развития почечной недостаточности [67].
Нарушение липидного метаболизма влияет также на темп потери почечной функции. По данным Helsinki Heart Study, снижение ЛПВП и повышение соотношения ЛПНП/ЛПВН у лиц среднего возраста с АГ приводило к более быстрому снижению СКФ [68]. В большинстве исследований, включивших больных СД, дислидемия наряду с АГ, гликированным гемоглобином, альбуминурией была фактором риска развития нефропатии [69, 70].
Значение нарушений липидного обмена в прогрессировании ХБП также подтверждено в ряде работ. В исследовании MDRD, включившем пациентов с умеренной и выраженной почечной недостаточностью, снижение уровня ЛПВП было независимым предиктором более быстрого снижения СКФ [71]. В другой работе показано влияние общего ХС, ХС ЛПНП и апоВ на ускорение темпа нарушения почечных функций [72]. У 104 больных ХБП, наблюдавшихся в среднем в течение 4 лет, значения ХС и ПУ были связаны с прогрессированием почечного поражения [73]. В то же время в итальянском исследовании, включившем 465 больных ХПН, уровень липидов не оказывал влияния на почечный исход [74]. В крупное исследование Early Treatment Diabetic Retinopathy Study были включены 2226 больных СД. За 5-летний период наблюдения 10,2% больных СД 1 типа и 9,8 % СД 2 типа потребовалось проведение заместительной почечной терапии. Среди факторов, приведших к тХПН, общими для всех типов СД были повышенный уровень ХС, уровень креатинина, низкий альбумин и анемия. Повышение значения ТГ было дополнительным фактором для проведения ЗПТ только при СД 2 типа [75].
Дислипидемия и сердечно-сосудистый риск при ХБП
У больных, получающих лечение ГД, связь между дислипидемией и сердечно-сосудистыми осложнениями не столь очевидна, как в общей популяции, где повышение уровня ЛПНП, ТГ и снижение ЛПВП - признанные факторы риска.
По данным ряда крупных исследований, связь между уровнем ХС и смертностью больных, леченных ГД, имеет U-образную форму [76, 77]. Так, при значениях ХС 200-250 мг/дл риск смертности был наименьший, тогда как при значениях ХС >350 мг/дл он возрастал в 1,3 раза, а при ХС < 100 мг/дл — в 4,2 раза. При дальнейшем статическом анализе на связь между низким уровнем ХС и смертностью оказывал влияние уровень альбумина плазмы крови. Схожие данные были получены в работе, включившей 1167 больных, находившихся на ГД, где наличие гипоальбуминемии и гипохолестеринемии коррелировало с высоким риском общей смертности. В то же время при уровне альбумина сыворотки > 4,5 г/дл высокий уровень ХС, как и в общей популяции, был ассоциирован с повышенной смертностью [78].
В CHOICE Study также показано парадоксальное влияние уровня дислипидемии в сочетании с системным воспалением и/или синдромом БЭН на выживаемость больных, получавших диализную терапию [79].
Таким образом, можно предположить, что обратная связь между уровнем холестерина и смертностью при уремии опосредована липидснижающим влиянием синдромов БЭН и/или системного воспаления.
В крупном японском исследовании (45 390 больных, находившихся на ГД) оценивали риск сердечно-сосудистых катастроф в развитии фатальных осложнений. При мультивариантном регрессионном анализе частота инфарктов миокарда положительно коррелировала с ХС не-ЛПВП и обратно с ХС ЛПВП. Частота ишемических инсультов была ассоциирована с ХС не-ЛПВП, тогда как между развитием геморрагического инсульта и липидными параметрами связь не выявлена. В то же время липидные показатели не оказывали влияния на смертность при развившихся осложнениях, но имели значение старший возраст, низкий ИМТ и высокий С-реактивный белок [80].
По нашим данным, при оценке влияния факторов риска на развитие атеросклероза у 165 больных со 2—4-й стадиями ХБП по мере ухудшения почечной функции (СКФ < 30 мл/ мин) мы выявили парадоксальную связь между уровнем общего ХС сыворотки крови и наличием атеросклеротического поражения сосудов (r = -0,32, р < 0,05). Кроме того, при корреляционном анализе развитие доклинической стадии атеросклероза, определяемое по толщине комплекса интима-медиа (ТИМ) сосудистой стенки в магистральных сосудах, было ассоциировано с нарушениями липидного обмена (табл. 3) [81].
Таблица 3. Корреляция толщины комплекса ТИМс факторами риска (n = 37).
.")
Примечание. н. д. — недостоверно.
**р<0,05, ТИМ ОСА и ОБА — толщина комплекса интима-медиа общей сонной артерии, общей бедренной артерии.
G.M. London (1990) выявил связь между скоростью пульсовой волны в аорте и уровнем ЛПВП у больных ТПН [82]. В иследовании HEMO Study, оценивавшем связь между “традиционными” факторами риска и наличием атеросклероза сосудов у 936 больных, находившихся на программном ГД, не удалось выявить связь между дислипидемией и поражением сосудов [83]. Схожие результаты в отношении исходов сердечно-сосудистых осложнений продемонстрированы в работе, включившей 840 больных с 3—4-й стадиями ХБП, когда наличие липидных нарушений не влияло на общую и сердечно-сосудистую смертность [84].
Частично это может объясняться тем, что высокий сердечно-сосудистый риск при почечной недостаточности кроме дислипидемии может быть связан с другими механизмами: анемией, кальцификацией сосудов, эндотелиальной дисфункцией, оксидативным стрессом, системным воспалением. Кроме того, при оценке атерогенного потенциала дислипидемии учитываются не все классы и не все характеристики липопротеинов. С учетом роли аполипопротеинов как переносчиков липидов, регуляторов ферментов липидного метаболизма и лигандов для связывания со специфическими рецепторами вероятен их вклад в развитие уремической дислипидемии. Продемонстрировано также, что сывороточный уровень ЛППП может являться предиктором атеросклеротического поражения аорты у больных, находящихся на программном ГД [85].
В исследовании, включившем 607 больных, получавших диализную терапию, была выявлена связь низкомолекулярных изоформ ЛП(а) с поражением коронарных артерий [86]. В проспективном 5-летнем исследовании 440 пациентов, леченных ГД, F. Kronenberg et al. (2000) выявили взаимосвязь между низкомолекулярным фенотипом ЛП(а) с выраженностью поражения коронарных сосудов [87].
В общей популяции у лиц, имевших ССЗ, уровень ароА-IV был ниже по сравнению со здоровым контролем независимо от концентрации ЛПВП и ТГ [88,89]. Похожие результаты были получены в Mild to Moderate Kidney Disease Study [90]. Уровень ароА-IV у больных с почечной недостаточностью и сердечно-сосудистыми осложнениями был ниже по сравнению с больными, не имевшими кардиоваскулярных осложнений.
Влияние дислипидемии на энергетический метаболизм при ХБП
Нарушение метаболизма липопротеинов, богатых ТГ, может оказывать неблагоприятный эффект не только на развитие сердечно-сосудистых заболеваний, но и на энергический метаболизм при ХПН [5]. Свободные жирные кислоты (СЖК) и глюкоза — основной источник энергии, обеспечивающий все механические, биохимические и биофизические функции организма. Снижение липолиза из-за дефицита липопротеинлипаз или рецепторов к ЛПОНП в скелетных мышцах и миокарде может нарушать поступление СЖК в эти ткани. Инсулинорезистентность, часто сопряженная с почечной недостаточностью, ограничивает доступность глюкозы для образования энергии в мышечной ткани. Все это, по мнению N.D. Vaziri, ограничивает возможность физической активности при ТПН [5]. Наряду с влиянием на мышечную ткань ХПН также приводит к дезрегуляции липопротеинлипаз и рецепторов ЛПОНП в жировой ткани. Так, в эксперименте на жировой ткани крыс с ХПН отмечено повышение образования СЖК и усиление экспрессии ферментов, участвующих в их синтезе [92, 93]. Считается, что это компенсаторный ответ сниженному поступлению СЖК в адипоциты вследствие дефицита рецепторов LPL и ЛПОНП [94]. Не исключено, что данный феномен частично может быть связан с потерей и недостатком веса при уремии [5]. Патогенетические и прогностическое значения нарушений липопротеидов при ХБП с точки зрения как прогрессирования почечного процесса, так и драматического нарастания риска сердечно-сосудистых осложнений определяют необходимость оптимизации соответствующей терапевтической стратегии.



{kind=link}
{kind=link}