
Результаты исследований, направленных на детализацию патогенеза вторичного гиперпаратиреоза при хронической болезни почек, в последние годы дополнились принципиально новыми данными. Установлены ранее не известные факторы, продуцирующиеся почечной и костной тканью, участвующие в регуляции гомеостаза фосфора, витамина D и минерализации костной ткани, – морфогенетические белки (FGF-23 и Klotho) [1, 2, 4]. В настоящее время поддержание постоянства физиологической плазменной концентрации фосфора рассматривается как результат комплексного взаимодействия паращитовидных желез (ПЩЖ), почек и костной ткани. При этом четко установлена активная эндокринная роль костной ткани [2–6].
При почечной недостаточности нарушаются все звенья минерального и костного обмена. Уже при снижении СКФ ниже 60 мл/мин/1,73 м2 уменьшается фильтрация фосфора и повышается его сывороточная концентрация, что вызывает повышение секреции FGF-23 и ПТГ; у детей аналогичные
изменения начинаются с ХБП II стадии [1, 4, 7].
Установлено, что уровень FGF-23 повышается в сыворотке крови при ХБП по мере снижения СКФ, значительно опережая увеличение сывороточной концентрации фосфора [4, 8, 9].
FGF-23 подавляют реабсорбцию фосфора, таким образом нормализуя его уровень в сыворотке крови, но при падении СКФ ниже 30 мл/мин/1,73 м2 этот механизм поддержания нормальной сывороточной концентрации фосфора становится недостаточно эффективным и развивается стойкая гиперфосфатемия, что стимулирует усиленную секрецию FGF-23 и в дальнейшем – ПТГ [1, 4, 7, 8].
Поскольку уровень сывороточного FGF-23 у больных ХБП часто предшествует гиперфосфатемии, резистентность к FGF-23 может быть одним из наиболее ранних проявлений нарушения метаболизма фосфора при ХБП [8]. Предполагают, что развитие резистентности к FGF-23 вызвано снижением
почечной экспрессии Klotho, который является кофактором действия FGF-23 [2, 4, 6, 10]. В связи с этим низкий уровень экспрессии трансмембранной формы белка Klotho в почках может быть фактором неблагоприятного отдаленного прогноза больных ХБП [2, 3, 6, 10–12].
Центральная роль FGF23/Klotho в механизмах развития ВГПТ в настоящее время является доказанной. С одной стороны, увеличение продукции FGF-23 начиная с ранних стадий ХБП препятствует развитию гиперфосфатемии и объясняет, почему сывороточная концентрация фосфора остается нормальной вплоть до выраженного снижения СКФ. С другой стороны, FGF-23 приводит к ингибированию образования кальцитриола и формированию ВГПТ за счет ослабления геномных механизмов контроля синтеза иПТГ [8, 13–15].
Получены подтверждения наличия в ПЩЖ внутриклеточных путей, общих для передачи различных внеклеточных сигналов, таких как изменения концентрации кальция, фосфора, 1,25(OH)2D3 и FGF-23/FGFR/Klotho, направленных и на регуляцию геномной экспрессии ПТГ, его секрецию, а
также на пролиферативные процессы в органе и механизмы пролиферации клеток ПЩЖ [16]. A.S. Dusso и соавт. (2001) показали, что на ранних стадиях ХБП ограничение потребления фосфора блокирует развитие гиперплазии ПЩЖ в результате специфической индукции мРНК и синтеза р21-ингибитора циклин-зависимой киназы. Напротив, диета с большим содержанием фосфора индуцировала трансформирующий фактор роста альфа (TGF-α – лиганд EGFR) и его конвертазу ADAM17 (асимметричный диметиларгинин 17), который, полагают, является аутокринным сигналом стимуляции клеточного роста и гиперплазии ПЩЖ [16–18].
Установлено, что ADAM17 может наряду с другими мембранными протеазами (BACE1, ADAM10) принимать участие в протеолизе белка Klotho [14]. При этом Klotho утрачивает свои рецепторные свойства, формируя развитие FGF-23-резистентности органов-мишеней. В то же время отщепленная
внеклеточная (экстрацеллюлярная) часть молекулы Klotho, попадая в циркуляцию, имеет не зависимые от FGF-23 плейотропные биологические эффекты в виде ингибирования Na-Р-транспортеров (NPT2a, NPT2c, NPT3), активации кальциевого и калиевого ионных каналов (TRPV5, TRPV6, ROMK1) [11,
19–21]. Активация ADAM17 связана и со снижением продукции кальцитриола при ВГПТ, поскольку VDR-активаторы блокируют его экспрессию наряду с инактивацией EGFR [22]. Следует также отметить, что активация ADAM17 в почке под действием ангиотензина II может приводить к эффектам, имеющим системное значение. ADAM17-опосредованное локальное высвобождение и увеличение в циркуляции TNF-α, обладающего известными профибротическими/провоспалительными свойствами, и молекул адгезии (ICAM-1 и VCAM-1), могут приводить к развитию системного воспалительного стресса и сердечно-сосудистых осложнений [24].
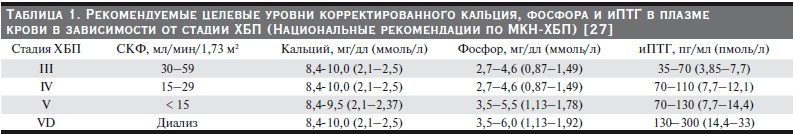
Таблица 1. Рекомендуемые целевые уровни корректированного кальция, фосфора и иПТГ в плазме
крови в зависимости от стадии ХБП (Национальные рекомендации по МКН-ХБП) [27].
Пролиферативные процессы в ПЩЖ также могут контролироваться Pit-1 – натрий-зависимым ко-транспортером фосфора, активность которого в свою очередь регулируется фосфором
и кальцитриолом. Недавно было показано, что снижение экспрессии Pit-1 вследствие нагрузки фосфором приводит к увеличению пролиферативной активности клеток ПЩЖ [23].
При ВГПТ под влиянием повышенной в плазме концентрации иПТГ в костной ткани активируются остеокласты, резорбирующие кость, образуя полости (лакунарная резорбция), которые заполняются остеобластами, синтезирующими коллаген. В результате этих процессов образуется неорганизованный
остеоид, после кальцификации которого формируется неорганизованная кость (women bone) со сниженной механической прочностью [25].
У пациентов с ВГПТ классическим поражением костной ткани является фиброзно-кистозный остеит, хотя последние исследования показали возможность развития при ВГПТ других форм костной патологии, таких как АБС [25].
Надежный диагностический критерий ВГПТ – повышение в сыворотке крови концентрации иПТГ. Диагноз вторичного гиперпаратиреоза ставится пациенту с ХПБ VD-стадии при уровне иПТГ > 300 пг/мл и/или выявлении почечной остеодистрофии [5, 6, 26]. Диагностическая ценность повышенного
иПТГ снижается при обнаружении алюминиевой перегрузки (50 мг/л/), т. к. в данном случае повышение иПТГ является компенсаторным [26, 27]. Для поддержания процесса ремоделирования кости на нормальном уровне содержание иПТГ у пациентов с ХБП должно быть в 2–3 раза выше, чем у здоровых лиц (табл. 1).
Определение биохимических маркеров формирования костной ткани пока не является частью рутинной диагностической программы исследования пациентов с МКН-ХБП, что связано с необходимостью использования сложных методик и дорогостоящего оборудования. В будущем они могут с пользой применяться для мониторинга течения болезни и эффективности лечения.
Инструментальная диагностика ВГПТ:
• Ультразвуковое исследование – наиболее простой и дешевый метод инструментальной диагностики образований ПЩЖ (УЗИ), может применяться интраоперационно для поиска желез. Результаты очень сильно зависят от опыта врача УЗ-диагностики, т. к. часто за ПЩЖ принимают узлы щитовидной железы, лимфатические узлы, мышцы, сосуды. УЗИ щитовидной железы информативно только
при расположении паратиреоаденом в типичных местах – в области щитовидной железы.
• Рентгенография позволяет обнаруживать остеопороз, кистозные изменения костей, патологические переломы. Для оценки плотности костной ткани проводится денситометрия. • Рентгенотомография загрудинного пространства с пищеводным контрастированием бариевой взвесью позволяет выявлять паратиреоаденому и ее местоположение.
• Компьютерная томография с внутривенным контрастированием – очень чувствительный метод исследования, его преимущество заключается в том, что можно выявлять загрудинные ПЩЖ.
• Магнитно-резонансная томография по информативности превосходит КТ и УЗИ, визуализирует любую локализацию околощитовидных желез.
• Радионуклидная диагностика позволяет оценивать функциональную активность образований ПЩЖ.
Ранняя профилактика ВГПТ.
На ранних стадиях формирования ВГПТ наиболее существенное значение имеет увеличение пула фосфора из-за дисбаланса между его поступлением с пищей и почечной экскрецией в условиях снижения СКФ. Поэтому основной стратегией в профилактике МКН у больных ХБП должна быть диета с низким содержанием фосфора, а при ее недостаточности – применение фосфатсвязывающих препаратов. Ограничение потребления фосфора следует начинать на самых ранних стадиях ХБП – еще когда СКФ выше 60 мл/мин/1,73 м2 [26, 27].
Результаты исследований последних лет свидетельствуют, что у больных ХБП и гиперфосфатемией (фосфор сыворотки – 6,5 мг/дл) риск смерти от сердечно-сосудистых заболеваний, включая ИБС, более высокий, чем у тех, у кого уровень фосфора ниже 6,5 мг/дл [6, 28, 29]. Поскольку концентрация FGF-23
(более раннего, чем ПТГ, маркера МКН) снижается при уменьшении гиперфосфатемии, терапевтической стратегией должна быть диета с низким содержанием фосфатов и применение
фосфатсвязывающих препаратов уже с ранней стадии ХБП. Принимая во внимание эти данные, контроль уровня фосфора сыворотки ниже 6,5 мг/дл становится важной терапевтической задачей для этих пациентов. Эти меры могут способствовать профилактике ВГПТ и ССО у больных ХБП [26, 27].
Диета и фосфатсвязывающие препараты
Диета с низким содержанием фосфора и/или применение препаратов, связывающих фосфор в ЖКТ, все чаще признаются важным терапевтическим подходом к предотвращению опасных для жизни осложнений у больных ХБП. Согласно рекомендациям KDIGO (2008), потребление фосфора следует
ограничивать до 800–1000 мг/сут (с коррекцией на пищевую потребность в белке), если уровень фосфора в сыворотке крови выше 4,6 мг/дл (1,49 ммоль/л) при III–IV стадиях ХБП и выше 5,5 мг/дл (1,78 ммоль/л) у диализных больных или содержание иПТГ в плазме превышает целевой уровень, определенный для соответствующей стадии ХБП. Если, несмотря на ограничение поступления фосфора с пищей не удается контролировать его уровень или иПТГ в пределах целевых значений, необходимо назначать препараты, связывающие фосфор в ЖКТ [27]. В России для коррекции гиперфосфатемии наиболее часто используют кальция карбонат и кальция ацетат. Способность связывать фосфор у кальция ацетата в 2 раза выше, чем у кальция карбоната. Длительный прием фосфатсвязывающих препаратов на основе солей кальция может вызывать гиперкальциемию, которая на фоне приема кальция карбоната бывает в 3,5 раза чаще, чем при использовании кальция ацетата, т. к. взаимодействие кальция карбоната с фосфором начинается при рН 5,0, когда растворимость кальция карбоната снижается. Фосфатсвязывающие препараты на основе кальция эффективно снижают концентрацию фосфора в сыворотке и могут использоваться в качестве начальной фосфатсвязывающей терапии. При этом суммарная доза элементарного кальция, используемого для связывания фосфора, поступающего с пищей, не должна превышать 1,5 г/сут [27].
Таблица 2. Доза севеламера гидрохлорида в зависимости от исходного уровня фосфора в сыворотке крови.
Фосфатсвязывающие препараты на основе кальция не должны применяться диализными больными с гиперкальциемией (корректированный общий кальций сыворотки выше 10,2 мг/дл [2,54 ммоль/л]) и в тех случаях, когда уровень иПТГ плазмы ниже 150 пг/мл (16,5 пмоль/л) при 2 последовательных
измерениях. Таким больным следует отдавать предпочтение фосфатсвязывающим препаратам, не содержащим кальций [26, 29].
Магния гидроокись и магния карбонат также снижают уровень фосфора в сыворотке крови, однако оценка их безопасности и эффективности в контролируемых клинических исследованиях не изучалась. Магнийсодержащие фосфатсвязывающие препараты эффективны в больших дозах, поэтому
при их назначении следует снижать содержания магния в диализирующем растворе. Кроме того, их применение нередко вызывает диарею, гиперкалиемию и гипермагниемию (H.H. Malluche, H. Mawad, 2002) [30]. Комбинированный препарат, содержащий магния карбонат и кальция карбонат, считается пищевой добавкой, поэтому он не оценивался экспертами Американской администрации по контролю качества пищевых продуктов и лекарств (FDA) (J.A. Coladonato, 2005) [31]. По мнению членов рабочей группы KDIGO (2008), применение магнийсодержащих препаратов обоснованно только при неэффективности всех других фосфатсвязывающих препаратов [26].
Выраженная токсичность ограничивает применение и алюминийсодержащих препаратов для связывания фосфора в ЖКТ. Исключением считают кратковременную терапию при неэффективности других средств контроля гиперфосфатемии (E. Ritz, 2005) [32].
В настоящее время в клинической практике для связывания в ЖКТ фосфора, поступающего с пищей, все более широкое применение находит севеламера гидрохлорид (ренагель) – синтетический препарат, не содержащий ни кальция, ни гидроокиси алюминия. Препарат показан для коррекции
гиперфосфатемии у диализных пациентов, при этом важно отметить, что он, не абсорбируясь в ЖКТ, снижает в крови содержание фосфора, иПТГ частично корригирует дислипидемию. В связи с отсутствием кальциемического эффекта отмечается безопасность комбинации ренагеля с активными
аналогами витамина D, а также его эффективность при кальцификации сосудов и мягких тканей. Показанием к назначению ренагеля считают повышение сывороточных уровней фосфора и корректированного общего кальция соответственно выше 5,5 и 10,2 мг/дл при снижении иПТГ ниже 150 пг/мл и развитии метастатической кальцификации. При длительном применении ренагеля может снижаться диуретический эффект фуросемида, развиваться гипохлоремический ацидоз, связанный с
потерей бикарбоната. Выбор дозы севеламера гидрохлорид в зависимости от исходного уровня фосфора в сыворотке крови представлен в табл. 2 [26].
Переход с фосфатсвязывающих препаратов на основе кальция на севеламера гидрохлорид осуществляют в эквивалентных дозах (мг/кг) сравнительно с препаратами на основе кальция.
Доза севеламера гидрохлорида может варьироваться от 1 до 5 таблеток по 800 мг за один прием пищи [26].
Положительное влияние севеламера на сердечно-сосудистый прогноз показано в нескольких клинических исследованиях и подтверждено результатами мета-анализа. Так, исследование
DCOR (Dialysis Clinical Outcomes Revisited) было посвящено сравнительному влиянию севеламера и других фосфатсвязывающих препаратов на параметры, оценивающие заболеваемость и смертность более чем 2000 больных, находившихся на регулярном гемодиализе. Несмотря на то что четких различий по эффективности севеламера и других фосфатсвязывающих препаратов с точки зрения динамики заболевания и риска летальности установить не удалось, в одном из повторных анализов результатов данного исследования было установлено, что благодаря использованию севеламера уменьшилась частота госпитализаций, связанных со всеми возможными причинами, а также продолжительность пребывания в стационаре (W.L. St. Peter и соавт., 2008) [33].
Мета-анализ 5 клинических исследований (M. Tonelli и соавт., 2007) [34], включивший 2429 пациентов (2103 из них – из исследования DCOR), продемонстрировал снижение риска смерти, достигнутое благодаря применению севеламера, по сравнению с кальцийсодержащими фосфатсвязывающими
препаратами.
В последнее время в целях связывания в ЖКТ фосфора используют лантана карбонат (Fosrenol). Препарат производится в таблетках по 500, 750 и 1000 мг. Начальная доза – 750 мг ежедневно во время или сразу после еды. Продолжительность лечения – до нормализации уровня фосфора (1,13–1,78 ммоль/л; 3,5–5,5 мг/дл). Максимальная доза препарата – 3750 мг/сут (на короткий период времени 5–7 дней). Однако лантан частично всасывается в кровоток и может аккумулироваться в
печени, легких, а также в костной ткани и вызывать снижение костеобразования (A. Bellasi и соавт., 2006) [35].
Прошли клинические испытания севеламера карбонат и Zerenex-фосфат-биндера, содержащего соединения неорганического железа. Предварительные данные свидетельствуют о том, что их эффективность связывать в ЖКТ фосфор не уступает солям кальция [36].
Комплекс мероприятий, направленных на профилактику МКН, следует продолжать в течение всего додиализного этапа ХБП, поскольку среди наблюдаемых нами больных, получавших выше указанную терапию на преддиализном этапе, в течение 1-го года лечения программным ГД достоверно реже,
чем среди больных, которым коррекция МКН начата одновременно с ГД, отмечалось формирование ВГПТ и развитие ССО [37–40].
Таблица 3. Эффекты современных групп препаратов, используемых для профилактики и лечения нарушений фосфорно-кальциевого обмена при ХБП.
Лечение ВГПТ: селективные активаторы VDR, кальцимиметики, паратиреоидэктомия
Регуляция продукции иПТГ и развитие ВГПТ связаны со многими факторами, действие которых в значительной степени независимо и суммируется по мере развития ХБП, – это фосфор, Ca/CaSR, 1,25(OH)2 D/VDR и FGF-23/ FGFR/Klotho. В связи с этим неоправданно ожидать адекватной профилактики ВГПТ при применении только одного метода терапии. Появляется все больше данных о преимуществах комбинированного лечения ВГПТ, включающего активаторы VDR (в особенности селективные) и кальцимиметики (исследования OPTIMA, ADVANCE). Комбинированный метод
терапии ВГПТ может позволить, с одной стороны, добиваться потенцирования лечебных эффектов за счет воздействия на разные молекулярные механизмы ВГПТ, с другой – избегать развития ССО [26].
Парикальцитол – селективный активатор VDR
Интересны результаты клинического исследования, проведенного D. Hansen и соавт. (2012), выявившего снижение уровня “уремического фактора – FGF-23” у 57 диализных пациентов с ВГПТ в результате лечения парикальцитолом [41], а в эксперименте получены данные об увеличении экспрессии нефропротективного фактора Кlothо в почках парикальцитолом [11], что позволяет рассматривать парикальцитол как препарат, обладающий потенциалом воздействовать на
патофизиологически активные факторы прогрессирования ХБП и тем самым улучшать почечный прогноз. Однако для подтверждения данной гипотезы необходимы дополнительные клинические исследования. Схожие результаты были получены и в нашем исследовании, проведенном на базе Первого МГМУ им. И.М. Сеченова в Клинике нефрологии внутренних и профессиональных болезней им. Е.М. Тареева [39, 40]. По результатам данного исследования было установлено, что применение парикальцитола на додиализных стадиях ХБП в исходе поражения почек в рамках системных заболеваний с развитием вторичного гиперпаратиреоза сопровождается не только нормализацией уровней интактного паратиреоидного гормона (иПТГ), но и достоверным снижением суточной
протеинурии и артериальной гипертензии.
В когортном исследовании [42], включившем 67 399 диализных пациентов с ВГПТ, смертность среди пациентов, у которых лечение гиперпаратиреоза проводилось парикальцитолом, была ниже, чем среди пациентов, получавших кальцитриол: соответственно 3417/19 031 и 6805/30 471 пациенто-лет (p< 0,001). При анализе по методу Kaplan–Maier более высокая выживаемость также отмечена среди больных, принимавших парикальцитол, чем среди тех, кто принимал кальцитриол (p < 0,001). В то же время более высокая (2-летняя) выживаемость отмечена среди больных, переведенных с кальцитриола на парикальцитол, чем при замене парикальцитола на кальцитриол (73 против 64 %, p = 0,04).
Кальцимиметики — препараты, способные эффективно подавлять секрецию иПТГ. Их применение больными ВГПТ нередко позволяет избегать паратиреоидэктомии.
Кальцимиметик цинакальцет вызывает изменения кальциевых рецепторов ПЩЖ, повышая их чувствительность к внеклеточному кальцию, что уменьшает продукцию и секрецию иПТГ,
может быть эффективным при гиперкальциемическом кризе. Начальная доза препарата составляет 30 мг/сут, ее обычно постепенно увеличивают до достижения концентрации иПТГ в сыворотке крови в пределах 200–300 пг/мл. Цинакальцет следует принимать один раз в сутки во время либо сразу после
еды. Стабильная концентрация препарата достигается в течение 7 дней. Переносимость препарата хорошая: только 5 % больных приходится прерывать лечение из-за побочных явлений (тошноты и рвоты). На фоне приема препарата может развиться гипокальциемия. При снижении корректированного общего кальция сыворотки крови менее 7,5 мг/дл следует назначать кальция карбонат и кальцитриол и снижать дозу цинакальцета или временно прекращать прием препарата до восстановления целевого уровня корректированного общего кальция. Высокая стоимость препарата ограничивает его широкое применение пациентами с терминальной ХБП. Также отсутствуют данные
о влиянии данного препарата на выживаемость пациентов. Сравнительная эффективность современных групп препаратов, используемых с целью профилактики и лечения нарушений фосфорно-кальциевого обмена при ХБП IV–V стадий, представлена в табл. 3 [26, 39,40].
Данные о роли комплекса асимметричный диметиларгинин 17/трансформирующий фактор роста-α /эндотелиальный фактор роста (ADAM17/TGF-α/EGFR), индуцируемого при активации рениннгиотензиновой системы (РАС) и дефиците кальцитриола, в структурной перестройке ПЩЖ [14, 10, 11] и в снижении экспрессии Klotho в почках [12,19,31,43], позволяют предполагать важность эффективной блокады РАС, коррекции дефицита D-гормона в профилактике и лечении ВГПТ.
Тактика ведения больных кальцифилаксией окончательно не определена. Местное лечение не эффективно, субтотальная паратиреоидэктомия у большинства больных приводила к регрессу синдрома кальцифилаксии, в части случаев повреждения кожи не заживали, а у некоторых они даже усугублялись [26, 44].
Паратиреоидэктомия. Лечение активными метаболитами витамина D не позволяет 60 % больных нормализовать уровень иПТГ, кальция и фосфора в крови, около 30 % больных резистентны к лечению метаболитами витамина D. Согласно Клиническим практическим рекомендациям KGIGO, больным тяжелым ВГПТ (сывороточный уровень иПТГ выше 800 пг/мл [88 пмоль/л]), ассоциированным с
неконтролируемой гиперкальциемией и/или гиперфосфатемией, показана паратиреоидэктомия [26, 45–47]. Другие показания – кальцийфилаксия, аденома одной из ПЩЖ или ее малигнизация. Этим пациентам может быть выполнена субтотальная паратиреоидэктомия или полная паратиреоидэктомия с аутотрансплантацией паратиреоидной ткани. Результативность и частота рецидивов сопоставимы как при субтотальной, так и при тотальной паратиреоидэктомии с аутотрансплантацией или без нее. Существует мнение, что тотальную паратиреоидэктомию не следует выполнять больным, которым планируется трансплантация почки, поскольку после пересадки контроль концентрации кальция в крови может быть затруднен [26].



{kind=link}
{kind=link}
{kind=link}